病毒清除验证指导原则及国内外申报差异
2019-04-13 佚名 生物制品圈

2017年,国家食品药品监督管理总局发布了《药品生产质量管理规范》生化药品附录的公告(2017年第29号),作为《药品生产质量管理规范(2010年修订)》配套文件。
生物药在我国已经形成“燎原之势”,各种生物仿制药、创新药的研发、生产不断地出现在市场上。根据《药品注册管理办法》的要求,由人的、动物的组织或者体液提取的制品、动物源性单克隆抗体及真核细胞表达的重组制品,尚需增加病毒灭活工艺验证资料。
2017年,国家食品药品监督管理总局发布了《药品生产质量管理规范》生化药品附录的公告(2017年第29号),作为《药品生产质量管理规范(2010年修订)》配套文件。
《生化药品附录》(以下简称《附录》)进一步对生化药品的范围进行了规范。生化药品是指从动物的器官、组织、体液、分泌物中经前处理、提取、分离、纯化等制得的安全、有效、质量可控的药品。主要包括:蛋白质、多肽、氨基酸及其衍生物、多糖、核苷酸及其衍生物、脂、酶及辅酶等。
《附录》的第五章部分对病毒去除/灭活及验证的必要性、适用性、有效性等提出具体的要求,明确基于风险控制原则,结合品种特性和原材料来源,采用有效去除/灭活病毒工艺步骤和方法。病毒去除/灭活的同时,不应对产品质量有不良影响。在生产工艺发生变更时,应重新评估病毒去除/灭活工艺的适用性。并且病毒去除/灭活验证不能代替原材料及生产过程的质量管理要求。
《附录》中提到“病毒去除/灭活工艺的有效性验证可参照有关病毒去除/灭活技术方法和指导原则等相关规定。”这里提到的“有关原则”大多数参考药品评审中心2007年发布的《生物组织提取制品和真核细胞表达制品的病毒安全性评价技术审评一般原则》(以下简称一般原则)以及2002年发布的《血液制品去除灭活病毒技术方法及验证指导原则》。
在《一般原则》中,指出了病毒安全性控制的迫切性以及适用范围,对病毒污染的可能来源进行分析,根据病毒的不同来源采取不同的控制和检测方法,并对病毒去除/灭活验证研究及有效工艺步骤的评价提出基本原则和方法。
在国内进行新药申报时提交的病毒清除验证以《生物组织提取制品和真核细胞表达制品的病毒安全性评价技术审评一般原则》及《血液制品去除/灭活病毒技术方法及验证指导原则》为主。如果需要在国外申报或中外双报,则需以《ViralSafety Evaluation of Biotechnology Products Derived from Cell Linesof Human or Animal Origin》、《Pointsto consider in the characterization of cell lines used to producebiologicals》、《Guidelineon Virus Safety Evaluation of biotechnological InvestigationalMedicinal Products》为主。
指导原则的不同必然造成国内外申报存在一些差异,具体主要有以下三个方面:
1、指示病毒的差异
国内的指导原则要求比较详细具体,明确指出了病毒的类型。规则要求:“一个典型的验证研究所选择的病毒,至少应包括单链和双链的RNA及DNA、脂包膜和非脂包膜、强和弱抵抗力、大和小颗粒等病毒;去除/灭活技术方面可根据采用的具体方法选择恰当的适宜病毒,例如SD法可选用脂包膜病毒,膜过滤法可选用粒径小的病毒,加热法可选用脂包膜和非脂包膜病毒,低pH孵放法可选用对理化因素比较耐受的指示病毒等。”
当然,选择指示病毒的核心需要以生物组织原材料、种子细胞或组织原材料匀浆、培养细胞结束时的混悬液中可能出现的污染病毒为主,结合能够用于评价验证效果的指示病毒的可获得性与相关培养试验条件进行合理选择。
国外规则没有写明病毒的具体要求,仅要求尽可能选择污染产品最相似的病毒进行验证,即特异性指示病毒;同时需要关注理化性质较宽的非特异性病毒。没有像CFDA中那样详细描述病毒的特点。
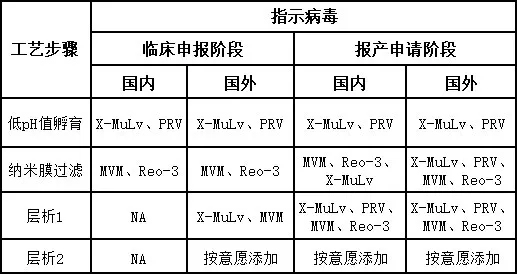
表.国内外申报中病毒选择差异
2、工艺选择上的差异
国内IND申报时,一般选择低pH和膜过滤这两个工艺。国外进行申报时,除了选择低pH和膜过滤这两个工艺,还需进行层析工艺。
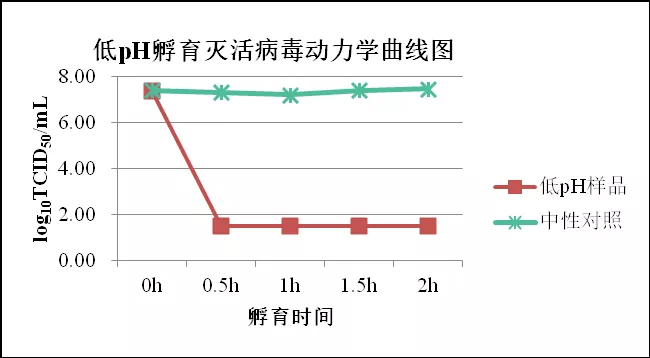
图.低pH孵育灭活病毒动力学曲线图
低pH处理法属于化学病毒去除/灭活的一种,原理是利用病毒表面的抗原在低pH值的条件下,电荷发生改变,蛋白质的空间结构也发生不可逆的改变,从而使病毒丧失与细胞受体结合的能力,阻止其侵染细胞。常用于单克隆抗体的病毒灭活工艺。一般选择3-3.7的pH处理2-24h。
膜过滤工艺是一种物理病毒去除/灭活方法,原理是选择孔径比病毒有效直径小的滤膜,对产品进行处理,将病毒与产品分离。膜过滤不能单独使用,需要与其他方法联合使用。在病毒验证时,需要综合考虑蛋白溶液的浓度、滤速、压力和过滤量等重要参数。
层析法最常用的是亲和层析和离子交换层析。层析法是目前常用的样品不同组分分离技术,利用各组分与固定相亲和力的差异或相互作用不同的原理,实现病毒与样品分离的目的。
3、在工艺重复性方面的差异
国内的指导原则虽然没有硬性的规定,但通常需要做三个批次。在国外的指导原则中,明确指出至少分别做两次独立的研究来证实清除的可重复性,因此一般是一批样品重复两次。
病毒去除/灭活工艺验证流程涉及多个方面,比如指示病毒的选择,验证方案的选择,清除效果的判定,以及统计处理分析等。
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言










#申报#
35