以双侧弥漫性脑白质病变为影像特征的大脑淋巴瘤病1例
2020-02-10 陶兢兢 张一望 黄振超 中国神经精神疾病杂志

大脑淋巴瘤病(lymphomatosis cerebri,LC)是原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)的一种罕见变异型。其特征为弥漫浸润性病变而不形成局部肿块及组织结构破坏,且影像学表现亦与PCNSL的不同,主要表现为双侧弥漫性脑白质病变。目前国内外关于LC的文献报道并不多,已报告的具有完善的影像学及病理学资料的病例
大脑淋巴瘤病(lymphomatosis cerebri,LC)是原发性中枢神经系统淋巴瘤(primary central nervous system lymphoma,PCNSL)的一种罕见变异型。其特征为弥漫浸润性病变而不形成局部肿块及组织结构破坏,且影像学表现亦与PCNSL的不同,主要表现为双侧弥漫性脑白质病变。目前国内外关于LC的文献报道并不多,已报告的具有完善的影像学及病理学资料的病例共63例。由于LC临床较为罕见,临床表现亦无特异性,故易被临床医生忽视而引起误诊或漏诊。本文报告我院收治的1例LC,并复习相关文献,主要对其临床表现、影像特点及病理表现进行探讨,以期提高临床医生对该病的重视和认识,降低漏诊或误诊率。
1.临床资料
1.1起病情况
患者,男,61岁,因“嗜睡、乏力2个月”于2018年7月6日入住我院。患者入院2个月前,无明显诱因下出现嗜睡、乏力,缄默状,少活动,无头痛头晕,无发热恶寒,无胸闷心慌等不适。遂由家属于6月4日送至当地医院就诊,查头颅MRI提示脑干、双侧颞叶、基底节区、右侧放射冠区梗死灶,诊断为脑梗死,予对症处理后患者症状未见缓解,并进行性加重,为进一步系统诊疗入住我院。患者既往糖尿病病史20余年,平素血糖控制不详。无艾滋、梅毒、乙肝、结核等传染病史。无免疫抑制剂药物服用史。无毒物接触史。无外伤手术史。无吸烟嗜酒史。无家族遗传史。
1.2体格检查
生命体征平稳,心肺腹部查体未见异常。专科查体:嗜睡,表情淡漠,缄默状,计算力、记忆力、理解力、定向力等检查不配合。双侧瞳孔等大等圆,直径约3mm,对光反射灵敏。鼻唇沟对称,伸舌不配合,咽反射正常。深浅感觉检查不配合。四肢肌张力增高,右侧肢体肌力3级,左侧肢体肌力4级,未见不自主运动,共济运动检查不配合,四肢腱反射减弱,双侧Babinski’s征(+)。脑膜刺激征(+)。
1.3辅助检查
1.3.1实验室检查
血常规:RBC:3.41×1012/L(4.3~5.8/L),Hb:107g/L(130~175g/L)。降钙素原:0.06ng/mL(0~0.05ng/mL)。超敏C-反应蛋白:17.63mg/L(0~6mg/L);肿瘤标志物:CEA:9.39ng/mL(0~5ng/mL);PSA3项:TPSA:5.02ng/mL(0~4ng/mL);细胞角蛋白19片段:3.9ng/mL(0.1~3.3ng/mL);血β2微球蛋白:2.59mg/L(0.7~1.8mg/L)。痰细菌培养:肺炎克雷伯菌,ESBL。脑脊液常规、生化检查结果见图1。

图1 脑脊液常规、生化检查结果
3次脑脊液液基细胞学检查均未见肿瘤细胞。脑脊液细菌培养、墨汁染色、自身免疫性脑炎相关抗体、寡克隆带均阴性。痰真菌、尿细菌培养阴性;痰抗酸染色找TB菌、血清结核抗体阴性。乙肝、梅毒、艾滋、肝肾功能、电解质、甲状腺功能、血管炎抗体、自身免疫抗体、凝血功能、其他肿瘤标志物及二便常规均未见异常。
1.3.2影像学检查
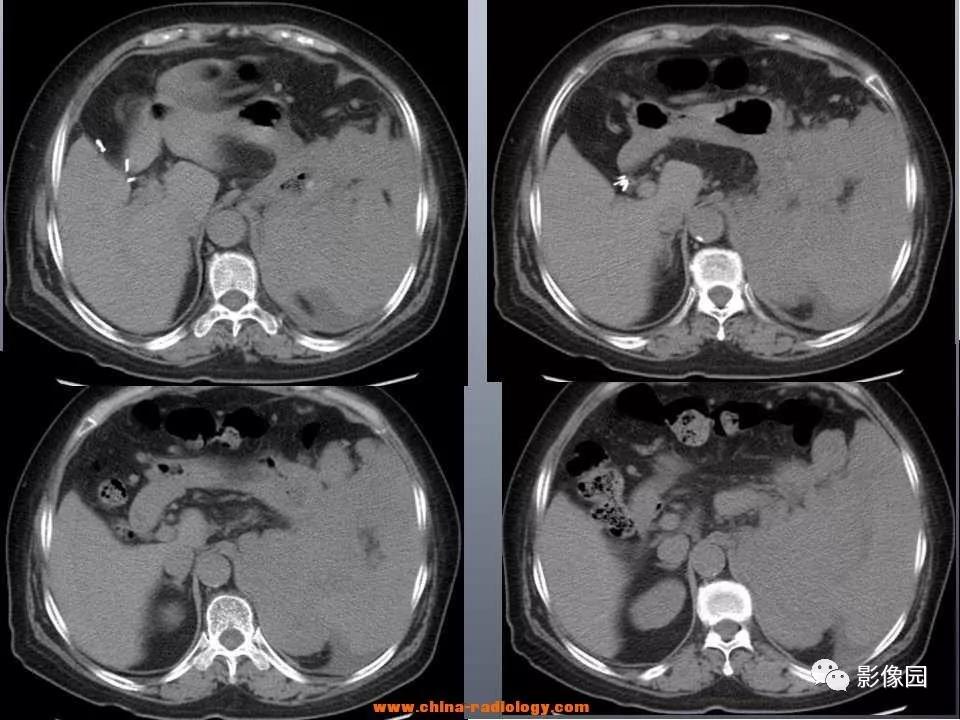
颅内外血管彩色B超检查无血管狭窄。6月28日头颅MRI平扫+DWI(见图2):双侧基底节区、丘脑,脑干、侧脑室前角、后角、两旁脑白质内,双侧颞叶,右侧半卵圆中心多发异常信号,建议进一步增强MRI后再诊。
7月5日头颅MRI增强(见图2):双侧基底节区、丘脑,脑干、侧脑室前角、后角、两旁脑白质内、双侧颞叶、右侧半卵圆中心多发异常信号,部分病灶明显强化,考虑感染性病变,请结合临床。7月17日PET/CT:①脑桥、双侧基底节、双侧丘脑、右侧颞叶、右侧半卵圆中心多发异常密度灶,代谢增高,以右侧丘脑-基底节-颞叶病灶为著,考虑非肿瘤性病变(感染性病变可能性大),并右侧颞叶局部水肿、代谢减低。②余全身未见明确异常高代谢病灶。
1.4诊断、治疗与随访
入院后考虑炎症可能性大,给予甲强龙1g/d冲击治疗,3d后逐渐改为口服,并结合抗感染、抗精神症状、营养神经等对症处理。激素冲击治疗后患者病情得到缓解,意识清楚,对答切题,表情丰富,四肢肌力基本恢复正常,7月13日复查头颅MRI平扫+MRS(见图2)提示病变范围较前略缩小。2d后病情再次加重,主要表现为认知功能障碍、神经精神症状及局灶性神经功能缺损,此时激素治疗效果欠佳。遂于7月20日转入外院,行脑组织病理检查明确诊断。

图2 患者头颅影像A~J:6月28日头颅MRI平扫+DWI,A、B:T1WI呈略低信号,C、D:T2WI高信号,G、H:部分病灶DWI呈略高信号,I、J:ADC呈稍高信号,E、F:FLAIR呈高信号。K~N:7月5日头颅MRI,K、L:MRI增强:部分病灶明显强化,M、N:FLAIR提示病灶较前略增多。O~S:7月13日头颅MRI+MRS,O~R:对比7月5日片,病灶较前范围略缩小,组织肿胀较前减轻。S:病灶区域呈Cho峰增高,Cr峰不显著,NAA峰略下降,Cho/Cr为1.8
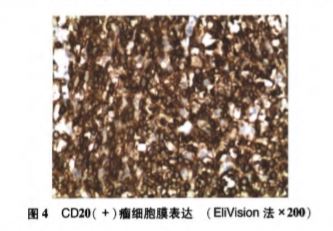
出院后电话随访,于8月3日行脑组织穿刺活检术。术后病理(见图3)提示白质内散在异型淋巴细胞弥漫性浸润,核仁明显,核分裂象易见。免疫组化结果:GFAP(+),CD3(少量+),CD20(+),CD79a(+),Ki-67(80%),S100(+),CD10(-),Mum-1(+),Bcl-2(+),Bcl-6(+),表1PCNSL和LC主要临床表现、影像学和组织病理学特征CD5(少量+),c-myc(散在+),CyclinD1(少量+)。结合临床及影像学表现考虑为B细胞来源的LC。病理确诊后患者家属拒绝接受进一步治疗,自动出院,2d后患者死亡。
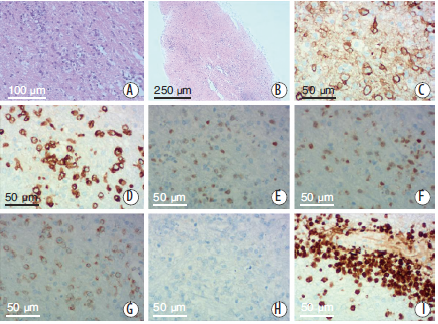
图3 患者活体脑组织病理检查结果(A×100,B×40)HE常规染色,见中等偏大的圆形细胞散在分布,细胞胞浆稀少,核染色质粗、核仁明显,核分裂象易见。免疫组化结果:CD20(C×200)、CD79a(D×200)染色阳性;CD10(H×200)染色阴性;Mum-1(E×200)、Bcl-6(F×200)染色阳性;Bcl-2(G×200)染色阳性;Ki-67(I×200)染色阳性,显示高增殖指数(80%)
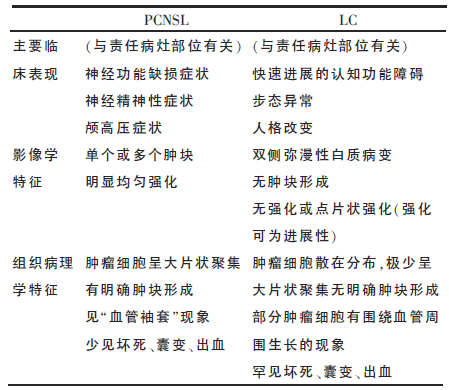
表1 PCNSL和LC主要临床表现、影像学和组织病理学特征
2.讨论
PCNSL是结外非霍奇金淋巴瘤的一种少见变异型,可累及脑、柔脑膜、眼或脊髓,而无全身性淋巴瘤的证据,约占原发性中枢神经系统肿瘤的4%。为了促进对PCNSL更精确的识别,已有研究者提出了几种类型:①以颅神经受累为特征的神经性淋巴瘤病,②淋巴瘤性脑膜炎-脑室炎,③主要表现为中风的血管内淋巴瘤,④LC。LC是PCNSL的一种罕见类型,其特征是淋巴细胞弥漫性浸润脑实质而不形成肿块或组织结构的破坏。其他类型的PCNSL多发生于免疫缺陷患者中,通常是慢性起病,而LC多发生于免疫功能正常患者中,多是亚急性起病。根据既往文献报道总结二者的主要差异,参见表1。
LC的临床表现多样且无特异性,与病灶侵犯的部位有关,最常见的症状是进行性认知功能下降或痴呆及步态异常,其他症状如局灶性神经功能缺损、人格改变、精神症状、行为改变、癫痫发作,以及头痛、呕吐等颅高压征,还有嗜睡乏力、视力障碍、食欲及体重下降等不常见的症状。LC的特征性影像学表现为双侧弥漫性脑白质病变,病位可涉及双侧大脑半球、脑室周围、基底节、丘脑或脑干,呈不完全对称性分布,一般增强无强化或点片状强化,亦可从无强化到出现强化、从轻度强化发展成斑片状或结节状或肿块样强化。在MRI上T1WI呈低或等信号,T2WI及FLAIR呈高信号,DWI及ADC呈稍高信号,其他类型的PCNSL通常在MRI上表现为单个或多个T2WI高信号且明显强化的肿块。
临床上与LC具有相似影像学特征的常见疾病有:中枢神经系统感染、炎症、中毒、代谢性疾病及自身免疫性脑炎、进行性多灶性白质脑病、脱髓鞘脑病、血管性白质脑病、大脑胶质瘤病等,极易引起误诊,其中大脑胶质瘤病的临床及影像学表现与LC皆相似,二者需通过病理活检鉴别;头颅MRS及PET/CT有助于鉴别肿瘤性与非肿瘤性疾病,但无特异性。
目前LC尚缺乏明确的临床诊断标准,仅根据临床及影像学资料诊断大脑淋巴瘤病比较困难,确诊仍需依据组织病理活检或尸体解剖。本例患者免疫功能正常,初期临床主要表现为嗜睡及人格改变,于当地医院就诊,查头颅MRI误诊为脑梗塞(但很遗憾当时未完善头颅MRI增强检查,该患者病灶早期的强化特点不明)。后期主要表现为认知功能障碍、人格改变和局灶性神经功能缺损。
患者的颅脑MRI特征为双侧大脑半球弥漫性白质病变,病灶T1WI呈略低信号,T2WI、FLAIR呈高信号,部分病灶DWI呈略高信号,ADC呈稍高信号,增强见部分病灶明显强化。根据患者的临床表现及影像学特点,难以鉴别脑白质病变及肿瘤性疾病。予激素治疗的同时,积极完善脑脊液细胞学检查、MRS及PET/CT等辅助鉴别诊断,但其结果均支持非肿瘤性病变。
脱髓鞘疾病、Binswanger病虽也具有与上述临床及影像学表现相似之处,但脱髓鞘疾病对激素治疗有效,且病程呈反复多发的特点,而本例患者激素治疗不敏感,且病程呈单一进展性,遂不考虑脱髓鞘疾病;Binswanger病属于脑小血管病,是在皮质下动脉硬化基础上,大脑半球白质弥漫性脱髓鞘性脑病,但起病缓慢,以慢性进行性痴呆为临床特征,故不考虑此病。其他如中毒性脑病、HIV脑病、代谢紊乱相关脑病、自身免疫性脑炎等,根据患者临床病史及实验室检查结果,亦不考虑。
总结该患者亚急性起病、病情进展快、激素治疗效果欠佳等临床特点,仍怀疑肿瘤可能性大,遂为患者行脑组织病理学检查以明确诊断。病理显示白质内见异常增生的淋巴细胞散在分布,染色质粗、核仁明显,核分裂象易见;CD20、CD79a染色阳性提示肿瘤细胞为B细胞来源;CD10染色阴性及Bcl-6、Mum-1染色阳性提示免疫分型为非生发中心型;Ki-67为80%,高度表达,提示肿瘤细胞的增殖极度活跃、预后差;结合免疫组化结果,考虑为高级别B细胞淋巴瘤,非生发中心型,最终依据病理结果结合临床及影像学表现明确诊断为LC。
与PCNSL可见明确肿块形成的病理表现不同,LC的病理学特征为圆形的肿瘤细胞沿着白质纤维束散在分布,而不会形成肿块,或偶见肿瘤细胞在血管周围聚集形成“血管袖套”现象,核分裂象易见,有核仁。其类型大多数为B细胞型,极少数为T细胞型。LC尚无标准的治疗方法,可参考PCNSL的治疗方法,文献报道有激素、化疗及放疗等治疗方法,多数情况下采用联合治疗方案。LC的预后较其他类型的PCNSL更差,大部分患者在3~12个月内死亡。由于患方因素,本例患者仅接受激素治疗而未接受放化疗,该患者病情进展快,病程短,预后差,与文献报道相符。
综上所述,临床医生、放射科医师和病理科医师必须了解PCNSL的LC类型。对于在MRI上呈双侧大脑半球弥漫性白质病变,同时累及深部脑白质,增强无强化或点片状强化,临床主要表现为认知功能障碍的患者,要高度怀疑为LC。及时识别这些磁共振成像特征可能有助于LC的早期诊断、早期治疗,改善预后。但最终明确诊断仍需通过组织病理学检查。值得注意的是,目前对于LC的研究主要来源于个案报道,只有少数病例完善了MRS及PET/CT检查,因此这些成像技术的诊断价值尚不明确,还有待进一步深入研究。
原始出处:
陶兢兢,张一望,黄振超,李惠平,王立新.以双侧弥漫性脑白质病变为影像特征的大脑淋巴瘤病1例[J].中国神经精神疾病杂志,2019,45(01):53-56.
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#大脑淋巴瘤病#
35
#白质#
42
#脑白质病变#
53
#弥漫性#
39
#双侧#
44
#白质病变#
52
#脑白质病#
43