病例分享:转氨酶升高、肝肿大待查1例——竟与罕见遗传性糖原累积病有关
2018-07-07 首都医科大学附属北京友谊医院肝病中心 武丽娜 王民 段维佳 赵新颜 王晓明 欧晓娟 贾继东 壹生
GSD是一种罕见的遗传性糖原代谢异常性疾病,其遗传方式大多数为常染色体隐性遗传,个别类型为伴X染色体遗传。由于糖原在转化为葡萄糖的代谢过程中出现异常,从而导致过多或异常糖原蓄积在肝脏及其他组织器官中。
病例介绍
患者男性,17岁。主因“发现转氨酶升高14年,间断意识丧失2年”于2017年8月14日入院。
现病史
14年前,患者出现发热、查转氨酶升高(具体不详),未接受特殊诊治。此后间断查转氨酶升高。
2年前,患者无明显诱因出现间断性意识丧失,伴有双上肢屈曲、双下肢伸直、牙关紧闭,无大小失禁,每次持续10~20分钟,可自行恢复。每年发作1~2次,患者未接受特殊诊治。
近半年,患者意识丧失发作较前频繁,约每2~3月发作1次,表现同前。就诊于当地医院。
查血常规大致正常。肝功能:丙氨酸氨基转移酶(ALT)133.9 U/L,天冬氨酸氨基转移酶(AST)181.9 U/L;碱性磷酸酶(ALP)257.9 U/L,γ-谷氨酰转肽酶(GGT)、总胆红素(TBIL)及白蛋白(ALB)正常;尿酸(UA)506 μmol/L,葡萄糖(GLU) 3.62 mmol/L。甲乙丙戊型肝炎病毒感染标志物均(-);免疫球蛋白及补体正常,抗核抗体(ANA)、抗线粒体抗体M2亚型(AMA-M2)(-);血清转铁蛋白饱和度、铁蛋白及铜蓝蛋白均正常。
心电图:窦性心律,左室肥厚,ST-T改变。
超声心动图:左房内径增大,左室壁增厚,少量心包积液,左室腔内高速血流信号。
脑电图:左侧额、中央、前颞、中颞可见中高幅尖慢波发放。
腹部超声:肝大,弥漫性肝病,脾大。
以“肝功能异常、肝肿大原因待查”收入我院进一步诊治。
既往史及家族史
左睾丸肿大2年。其外祖父逝于肝硬化(具体病因不详),其兄体检发现肝肿大(未明确诊断)。
入院时体格检查
全身皮肤巩膜无黄染,颈胸部未见蜘蛛痣,亦未见肝掌。全身浅表淋巴结未触及肿大。双肺呼吸音粗,未闻及干湿音及胸膜摩擦音。心前区无异常隆起,心率93次/分,律不齐,二尖瓣听诊区可闻及2~3级收缩期杂音。
腹平软,无腹壁静脉曲张,未见胃肠型及蠕动波;左上腹压痛,无反跳痛及肌紧张,墨菲征阴性;肝剑突下3 cm,肋下10 cm,边缘光滑,质软,肝区叩击痛阳性;脾肋下未及;移动性浊音阴性;肠鸣音4次/分。双下肢轻度水肿。左睾丸肿大,阴茎发育不良,第二性征不明显。
第一次分析讨论:疾病鉴别诊断
患者青年男性,幼年起病;肝脏方面表现为肝脏肿大、肝功能异常(ALT、AST、ALP升高),乙肝五项、丙肝抗体、铁代谢指标、铜兰蛋白、免疫球蛋白、AMA-M2、ANA大致正常,肝脏超声检查示肝弥漫性病变。目前诊断肝功能异常、肝脏肿大原因待查。
除肝脏外,病变还累及心脏、神经以及生殖系统,表现为左房内径增大,左室壁增厚、心律不齐;间断癫痫发作,近期更加频繁;身体发育迟缓、阴茎发育不良,第二性征不明显。结合患者家族史,考虑遗传代谢性肝病可能性大,例如肝豆状核变性、糖原累积病(GSD)等,亦不排除全身性疾病累及肝脏,例如淀粉样变性、结节病等。需要与下列疾病相鉴别。
肝豆状核变性 肝豆状核变性多发生于儿童及青少年时期,为常染色体隐性遗传性疾病。致病基因ATP7B基因突变导致三磷酸腺苷(ATP)酶功能减弱或消失,引起铜蓝蛋白合成减少以及胆道排铜障碍。
蓄积至体内的铜离子在肝、脑、肾、角膜等处沉积,引起进行性加重的肝硬化、锥体外系症状、精神症状、肾损害及角膜色素环(K-F环)等。可出现血清铜蓝蛋白降低、尿铜增加、角膜出现K-F环等铜代谢异常的证据。
本例患者青年男性,幼年起病,多系统受累,以肝脏及神经系统受累为突出表现,故需考虑此病可能。但外院化验铜蓝蛋白正常,需复查铜蓝蛋白,并进一步完善头颅核磁、眼裂隙灯检查、肝穿刺病理等检查,以进一步排除肝豆状核变性。
糖原累积病 GSD为少见的常染色体或伴X染色体的遗传性疾病,此病有很多类型,其中Ⅰ、Ⅲ、Ⅵ、Ⅸ型以肝脏病变为主,Ⅱ、Ⅴ、Ⅶ型以肌肉组织受损为主。主要表现为肝肿大、低血糖、高脂血症、高尿酸血症、生长发育迟缓、肌肉萎缩、肌张力低下、运动障碍,心室壁增厚、心脏扩大、心律失常等。
本例患者肝功能异常、肝肿大,伴有尿酸增高、发育迟缓,故不排除GSD。建议行肝穿刺活检明确诊断。
淀粉样变性 淀粉样变性为多种原因造成的淀粉样物质在体内各脏器沉积,可出现多系统受累。肝脏受累主要表现为肝肿大、ALP升高,可导致门脉高压进而出现食管静脉曲张和腹水。心脏受累常见,表现为心脏扩大、难治性心衰及心律失常。肾脏累及可表现为全身水肿、低蛋白血症、蛋白尿;胃肠道淀粉样变性可引起食管动力异常,胃张力迟缓,肠蠕动减慢,消化不良等。确诊主要依靠皮肤或肝脏等器官活检病理学检查。
本例患者虽然有肝肿大,但是生化指标以ALT和AST升高为主,与此病特点不符合,需进一步完善肝穿刺病理活检以排除诊断。
入院检查
实验室及影像学检查
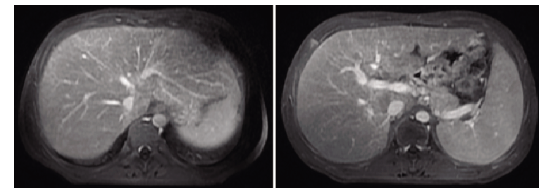
腹部增强MRI:患者腹部增强核磁显示肝脏体积增大,外形光滑;肝实质信号在T2WI上不均匀增高,可见多发细条索影,延迟强化,提示肝内少许纤维化改变;肝Glisson鞘稍增厚,少量腹水,脾大,符合慢性肝病表现。
血常规:白细胞(WBC)3.44×109/L,血红蛋白(Hb)116 g/L,血小板(PLT)123×109/L。
尿常规: 酮体(KET)(-),隐血(BLD)(-),尿蛋白(PRO)(-)。
肝功能:ALT 152 U/L,AST 218 U/L,ALP 304 U/L,GGT 82 g/L,TBIL 11.6 μmol/L,直接胆红素(DBIL) 2.6 μmol/L,ALB 31 g/L,GLU 3.6 mmol/L。
肾功能:肌酐(Cr) 57 μmol/L,尿素氮(BUN) 3.9 mmol/L,UA 491μmol/L。
血脂:胆固醇(CHOL)3.23 mmol/L,甘油三酯(TG)1.28 mmol/L。凝血酶原活动度(PTA) 87%。红细胞沉降率(ESR)16 mm/h,C反应蛋白(CRP) 4 mg/L。
肌酶:肌酸激酶(CK)1662 U/L,肌酸激酶同工酶(CK-MB)83.80 ng/ml,心肌肌钙蛋白I(cTnI)、心肌肌钙蛋白T(cTnT)、N端脑钠肽前体(NT-proBNP)均正常。
甲状腺功能、性激素、生长激素均正常。眼科会诊未见角膜K-F环。
腹部增强磁共振成像(MRI):肝肿大、少量腹水,脾大(图1)。
脑电图:全导可见弥漫性频繁发作的低中幅慢波,背景生理波调节差,左顶部α波减少。头颅MRI:双侧枕叶和左侧额叶局部脑萎缩改变,双侧枕叶深部白质脱髓鞘改变。
泌尿系统超声检查:双肾大小、回声正常,右附睾头囊肿;左侧精索静脉曲张,左侧精索鞘膜积液;左侧精索静脉旁囊性结节。
于2017年8月16日,在超声引导下行肝穿刺活检,送病理学检查。
病理学结果

图2 患者肝脏穿刺病理检查结果
肝穿病理可见小叶内肝细胞普遍肿大,部分肝细胞体积是正常肝细胞体积的 2~3 倍,多数肝细胞疏松水肿,胞质淡染,细胞膜突出,肿大肝细胞挤压肝窦致肝窦消失[图2-A,淀粉酶消化法(糖原D-PAS 染色)],过碘酸希夫(PAS)染色阳性物质(图2-B,PAS染色)经淀粉酶消化后消失(图2-C,糖原D-PAS染色),糖原核肝细胞易见。小叶内散在小坏死灶,未见泡沫样枯否细胞;未见明显脂变及窦周纤维化。
汇管区轻度扩大,以单核细胞及淋巴细胞为主的炎性浸润(图2-D,HE染色),轻度局灶界面炎;汇管区小胆管上皮增生、排列不整,门脉小支未见内皮炎,汇管区周边轻度细胆管反应,Masson染色提示汇管区间质轻度增生。
铜染色阴性,铁染色阴性。免疫组化染色:Mum-1(-)、CK7胆管上皮+、CK19胆管上皮+、CD38-、泛素细胞浆+。
病理诊断:符合糖原累积病(GSD)。
第二次分析讨论:鉴别糖原累积病(GSD)的具体分型
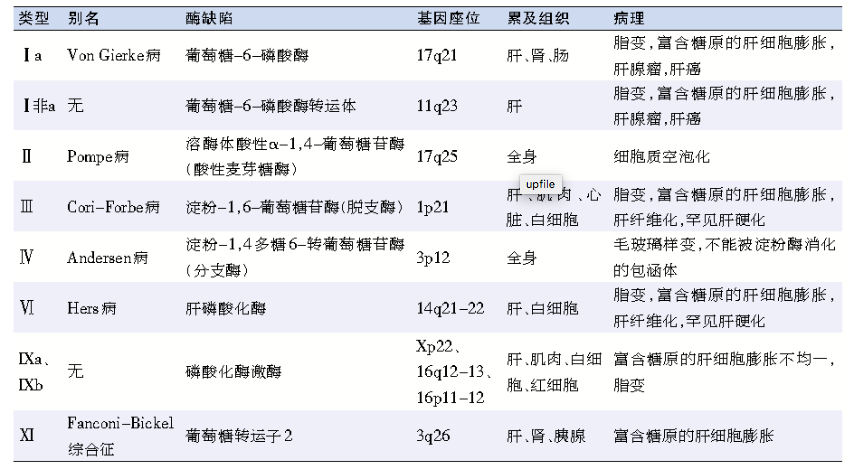
表 肝糖原累积病的主要分型及特点
患者青年男性,幼年发病,主要表现为肝脏受累(肿大、肝功能异常,以转氨酶升高为主,无肝腺瘤或肝癌),骨骼肌受累(肌酸激酶升高),心肌受累(左心室心肌肥大,左心室扩大),中枢神经系统受累[反复癫痫发作,脑电图及头颅磁共振成像(MRI)异常],以及身体迟缓、第二性征不明显。但患者血液生化仅表现为血尿酸轻度升高,无低血糖发作,血脂正常,无代谢性酸中毒,无铁缺乏,无肾脏受累(肾功能正常,肾脏不大)。
肝脏病理所见肝细胞明显肿大,呈植物细胞样改变(假细胞壁),胞浆疏松淡染,未见明显脂变及窦周纤维化。PAS染色阳性物质经淀粉酶消化后消失。
综合患者的临床表现、家族史和肝脏病理学特征,主要考虑GSD。根据酶缺陷或转运体的不同,GSD可分为十几个类型,其中Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅵ、Ⅸ、Ⅺ型主要累及肝脏,故称为肝糖原累积病,其中不同的分型又各具特点(表)。
Ⅲ型GSD为糖原脱支酶活性缺乏导致糖原支链不能被分解,使大量带短支链的形态结构异常的界限糊精在患者肝脏和(或)骨骼肌、心肌中堆积。表现为患儿自幼出现肝肿大,反复发生空腹低血糖,伴或不伴轻度无力;青春期后肝肿大减轻,低血糖发作减轻,但肌病表现逐渐明显,骨骼肌病变进行性加重,在生命后期进展为严重的肌无力。多数成人患者的心肌病临床症状并不明显,但有心电图和超声心动图异常,尤以左心室肥厚比较常见。
综合该患者临床表现、家族史和肝脏病理学特征,可明确GSD的诊断;结合病史及临床特点,首先考虑Ⅲ型GSD可能性大,但患者未进行基因检测和酶学测定,无法最终确定GSD具体分型。后续将进一步完善患者及其家属的基因检测,确定分型。
专家点评
GSD发病机制
GSD是一种罕见的遗传性糖原代谢异常性疾病,其遗传方式大多数为常染色体隐性遗传,个别类型为伴X染色体遗传。由于糖原在转化为葡萄糖的代谢过程中出现异常,从而导致过多或异常糖原蓄积在肝脏及其他组织器官中。
GSD诊断
诊断GSD需要综合患者的临床表现。例如肝脏受累(肝脏肿大,肝功能异常),常伴代谢异常(如空腹低血糖、高脂血症、高尿酸血症、高乳酸血症及酮症等)及生长发育迟缓等。骨骼肌受累可表现为肌无力、肌痛、肌痉挛、肌溶解等。心肌受累常表现为心室壁增厚、心脏扩大、心律失常、心衰等。
部分类型的患者可有肾脏受累(肾功能异常、肾脏肿大),以及粒细胞减少、反复感染、炎症性肠病等表现。对于怀疑GSD的患者需要完善肝、骨骼肌等受累脏器的穿刺活检,有条件的医疗机构可以进一步完善酶活性的检测及基因检测。
GSD分型
根据酶缺陷或转运体的不同,GSD可分为十几个类型。其中Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅵ、Ⅸ、Ⅺ型GSD主要累及肝脏,称为肝GSD。具体临床表现在第二次分析讨论中已有具体阐述。
累及肌肉的GSD以Ⅱ型、Ⅲ型、Ⅴ型病例较多。GSD的骨骼肌受累在临床上常表现为运动相关症状(运动不耐受,运动相关的肌痛、肌痉挛、反复发作运动诱发的急性肌球蛋白尿/横纹肌溶解,常见于Ⅴ、Ⅶ~Ⅻ型GSD),以及持续的进行性肌无力(常见于Ⅱ、Ⅲ、Ⅳ型 GSD)。
GSD治疗
饮食治疗 治疗方面,生玉米淀粉可缓慢释放葡萄糖,既能避免发生低血糖,亦能限制膳食中过剩的葡萄糖合成为糖原而过度沉积于肝脏。对于婴幼儿,白天可通过口服生玉米淀粉进行频繁的喂养,夜间可持续鼻饲或静脉点滴葡萄糖。对于年长儿,每2~3小时给予生玉米淀粉进行频繁的热量补充通常有效。
肝移植 肝移植可延长以肝硬化为主的GSD患者的寿命,但并不能缓解肝外其他组织器官的损害。GSDⅣ型患者肝硬化进展速度较快,对于病变仅限于肝脏的患者,需在5岁前进行肝移植。
GSDⅠ型及Ⅲ型患者可出现肝腺瘤,而肝腺瘤恶变是 GSD患者接受肝移植手术的适应证。但对于GSDⅠ型患者,肝移植无法改变肾脏疾病的病情;而Ⅲ型GSD患者病变弥散,很少有患者能从肝移植中获益。
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







#遗传性#
40
#肝肿大#
41
#转氨酶#
40
#罕见#
40
学习了很有用不错
76
了解一下.谢谢分享!
78
谢谢分享学习了
78