遗传病科普:苯丙酮尿症
2016-12-29 娓娓道来 中科基因生物科技

疾病简介苯丙酮尿症(phenylketonuria,PKU)是一种常染色体隐性遗传疾病,是先天性氨基酸代谢障碍中最为常见的一种,由于PAH缺乏,不能将苯丙氨酸转化为酪氨酸,致使苯丙氨酸在血液、脑脊液、各种组织中的浓度增高,同时由于主要代谢途径受阻,次要代谢途径增强,即在转氨酶作用下,苯丙氨酸脱氨基产生大量的苯丙酮酸,经氧化作用生成苯乙酸、苯乳酸和对羟基苯丙酮酸等旁路代谢产物,并自尿中大量排出。高浓
疾病简介
苯丙酮尿症(phenylketonuria,PKU)是一种常染色体隐性遗传疾病,是先天性氨基酸代谢障碍中最为常见的一种,由于PAH缺乏,不能将苯丙氨酸转化为酪氨酸,致使苯丙氨酸在血液、脑脊液、各种组织中的浓度增高,同时由于主要代谢途径受阻,次要代谢途径增强,即在转氨酶作用下,苯丙氨酸脱氨基产生大量的苯丙酮酸,经氧化作用生成苯乙酸、苯乳酸和对羟基苯丙酮酸等旁路代谢产物,并自尿中大量排出。高浓度的苯丙氨酸及其旁路代谢产物在脑组织中大量蓄积,导致脑细胞受损。本病因患儿尿液中排出大量苯丙酮酸代谢产物而得名,发病率具有种族和地域差异,我国的发病率总体为1:11000,北方人群高于南方人群。人类苯丙氨酸羟化酶基因位于第12号染色体上(12q22~12q24),基因全长约90Kb,有13个外显子和12个内含子,成熟的mRNA约2.4kb,编码451个氨基酸。通过对PKU病人进行基因分析,在中国人群中已发现了100种以上基因突变。根据报道,PAH基因突变位置多变,类型多样,呈现高度异质性,即不同种族和地区人群之间突变部位及分布差异很大。
PKU是为数不多的可治性的遗传病之一,早期诊断、早期治疗是其预后的关键。大规模的新生儿代谢病筛查为其早期发现、早期诊断及早期治疗提供了重要手段。
临床诊断
由于全国PKU新生儿疾病筛查的大力推广,目前较多患儿在新生儿期检出,患者多无临床表现。
一、临床表现
患儿出生时正常,通常在 3~6 个月时始出现症状,1 岁时症状明显,表现为:
1. 神经系统智力发育落后最为突出,智商常低于正常。有行为异常,如兴奋不安、忧郁、多动、孤僻等。可有癫痫小发作,少数呈现肌张力增高和腱反射亢进。
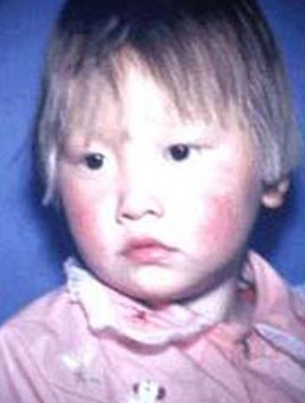
2. 皮肤患儿在出生数月后因黑色素合成不足,头发由黑变黄,皮肤白皙。皮肤湿疹较常见。
3. 体味由于尿和汗液中排出较多苯乙酸,身上有明显鼠尿臭味。
4. PKU母亲在未控制血苯丙氨酸浓度的情况下怀孕,其子女即使不是PKU,也常伴有小脑畸形和智力低下。
二、诊断方法和诊断步骤
新生儿疾病筛查
PKU是一种可治性的遗传性代谢病,其条件是要早诊断、早治疗。目前基本普及的新生儿疾病筛查系统,要求新生儿出生72小时并且充分哺乳后,取足跟血滴于S&S903新生儿筛查滤纸片上,室温自然干燥后装入塑料袋,置4度冰箱保存,递送新生儿筛查中心检测。荧光测定法或串联质谱测定凡初筛血苯丙氨酸浓度≥0.12mmol/L(2mg/dl),经原标本复查后大于切割值,通知招回采血。若血苯丙氨酸浓度仍大于切割值,转诊临床专科进一步诊断。如Phe浓度大于切割值,进一步鉴别诊断和确诊。患者在出生后2-3周开始得到治疗,预后良好。苯丙氨酸浓度测定正常浓度小于120μmol/L(2mg/dl),经典型PKU>1200μmol/L。
2. 尿蝶呤图谱分析血苯丙氨酸增高,除了有苯丙氨酸羟化酶活性降低外,还必须要与辅酶四氢生物蝶呤(tetrabiopterin, BH4)缺乏症鉴别,人体内的BH4 来源于三磷酸鸟苷(GTP), 在其合成和再生途径中必须经过三磷酸鸟苷环化水解酶 (GTPCH)、6-丙酮酰四氢蝶呤合成酶 (PTPS) 和二氢生物蝶啶还原酶 (DHPR) 的催化。BH4缺乏症的发生率占高苯丙氨酸血症的10%-15%,诊断主要依靠尿蝶呤谱分析。
尿蝶呤谱分析应用高效液相层析 (HPLC) 测定尿中新蝶呤(N)和生物蝶呤(B)。如因6-丙酮酰四氢蝶啶合成酶缺乏所致的BH4缺乏症,尿中新蝶呤明显增加,生物蝶呤下降,N/B增高,比值(B/B+N%) <10%。三磷酸鸟苷环化水解酶缺乏的患儿呈现蝶呤总排出量减少。
3. 干纸片法测定红细胞二氢喋呤还原酶。
4. 四氢生物蝶呤负荷试验在血苯丙氨酸浓度较高(>600mmol/L)情况下,直接给予口服BH4片20mg/Kg,BH4服前,服后2、4、6、8、24小时分别取血作Phe测定。对于血苯丙氨酸浓度<600mmol/L者,可作苯丙氨酸-四氢生物蝶呤联合负荷试验,即给患儿先口服苯丙氨酸(100mg/kg),服后3小时再口服BH4。四氢生物蝶呤负荷试验主要鉴别患者是否对四氢生物蝶呤负荷有反应,在服用BH4后24 h内,其血Phe浓度下降超过30%为有反应,见于四氢生物蝶呤缺乏症和部分PKU患者,后者称为四氢生物蝶呤反应性苯丙氨酸羟化酶缺乏症(BH4反应性PAH缺乏症)。
5. DNA分析对苯丙氨酸羟化酶可进行基因突变检测,对胎儿进行产前诊断。
三、 治疗
成年女性患者在怀孕前应重新开始饮食控制,血苯丙氨酸应该在300μmol/L以下,直至分娩,以免高苯丙氨酸血症影响胎儿。
产前基因诊断的策略
产前诊断是在对先证者和父母进行分析、确定其突变后,通过胎儿材料(绒毛、羊水)的DNA分析,有针对性地检测胎儿是否存在与先证者相同的基因突变。因此在对PKU高危家系进行产前诊断之前,必须明确先证者PAH基因的致病突变。
在产前诊断时,应联合应用AS-PCR和 STR 多态性分析,互相印证。如果出现不符合,尤其在只检测到母源突变时,可能提示胎儿材料有母源污染,甚至没有获得胎儿的材料而只是孕妇的材料。
在解释产前诊断结果时要说明诊断的准确性;采用什么途径进行产前诊断,取决于是否进行了预分析。
1.已经进行过预分析的病例:产前诊断时,针对性地检测家系中已经明确的突变和提供信息的多态性标记位点进行平行检测,结果互相印证。
2.如果没有进行过预先的基因分析:应在签署知情同意书时向家属明确告知,本例产前诊断服务的预期结果不确定性,并且只能针对 PAH基因进行分析。诊断中,应用 AS-PCR 检测常见 7 种 PAH突变,并用 PAH基因连锁的 STR多态性标记进行连锁分析。若能确定先证者两个等位基因突变,则结果明确;若基因突变不明确,胎儿又获得与先证者相同的多态性标记,则胎儿有风险。对于分析的结果及时与家长联系,确定是否深入分析。若要明确诊断,需要对 PAH基因进行全程测序。如果没有检测到 PAH 基因的突变,需要进行PTPS基因的序列测定,需要与家属联系。
产前诊断的技术方法
基因突变直接诊断:对PAH基因进行DNA序列分析是PKU基因诊断的金标准。在实际工作中主要先进行带有常见突变的外显子片段PCR扩展和DNA序列分析,再进行少见突变外显子的突变检测。PCR技术和分子杂交技术的结合(基因芯片)可对PAH基因突变进行快速诊断,有推广应用需求。
基因连锁间接诊断:在人PAH基因内含子3,距外显子约700bp位置发现[TCTA]n的短重复序列(STR),其重复单位在人群中具有长度多态性,PCR扩增产物长度为228-260bp不等,STR标记的杂合子频率较高,可快速对PKU基因提供产前诊断信息。
由于STR标记多态性达不到100%,必须联合基因突变检测,才可能进一步提高PKU的产前诊断率。
除了肝PAH活性缺如或降低可引起PKU外,还有PAH的辅因子发生变化也可引起。参与PAH作用的主要辅因子有5,6,7,8-四氢生物蝶呤(5,6,7,8-tetrahydrobiopterin),此物质为苯丙氨酸、酪氨酸和色氨酸羟化必须的辅因子。负责编码此物质的基因为6-丙酮酰四氢蝶呤合酶(6-pyruvoyltetrahydropterin)合成酶(PTPS)基因。如果这种酶基因发生突变,则PTP缺乏,PAH活性即使正常,也可引起PKU。另一种可引起PKU的酶是二氢蝶呤还原酶(dihydrofolate reductase)。据此,PKU的发病牵涉到至少3种酶基因,其中一个基因发生突变即可引起PAH活性缺如或减低,从而引起PKU。
四氢生物蝶呤(BH4)缺乏症又称非经典型PKU或恶性PKU,由于PAH辅助因子BH4缺乏所致。患儿除了有典型PKU表现外,神经系统(Nervous System)表现较为突出,如躯干肌张力下降,四肢肌张力增高,不自主运动,震颤,阵发性角弓反张,顽固性惊厥发作等。1%~3%患儿于1岁时发生严重的脑损害。BH4缺乏症者单独用低苯丙氨酸饮食治疗可使血苯丙氨酸浓度下降,但神经系统(Nervous System)的症状仍呈持续性进展。该病的发生率占PKU的10%左右,因此对所有高苯丙氨酸血症都应进行常规鉴别诊断【下一次介绍】。
遗传咨询
遗传咨询是一个与患者或者有风险个体的交流过程,其目的是帮助他们了解所患疾病的诊断、性质、发生和传递的可能性、可供选择的治疗或预防的措施,供患者及其家属在决定婚姻、生育等问题时参考。
PKU的遗传方式为常染色体隐性遗传,先证者父母为携带者,即携带有一个PKU致病突变位点的杂合子。携带者无PKU临床症状。先证者的同胞有1/4的风险患病,1/2的风险成为PKU致病突变携带者,并有1/4的可能性为不带有PKU致病突变的正常人。PKU家庭若要再次生育,要有基因检测的保证,需进行产前基因诊断。
先证者的后代肯定为携带者。若携带者频率为1/50,PKU患者的后代为患者的可能性为1/100。先证者父母的同胞为携带者的可能性为1/2。
如果产前诊断胎儿为患病者,应告知疾病危害性,处理应由患者家庭自主决定。
原始出处:
1. Goltsov AA, Eisensmith RC, Naughton ER, Jin L, Chakraborty R, Woo SL. A single polymorphic STR system in the human phenylalanine hydroxylase gene permits rapid prenatal diagnosis and carrier screening for phenylketonuria. Hum Mol Genet. 1993; 2(5): 577-581.
2. Eisensmith RC, Goltsov AA, Woo SL.A simple,rapid,and highly information PCR-based procedure for prenatal diagnosis and carrier screening of phenylketonuria. Prenat Diag. 1994; 14: 11131-8.
3. Goltsov AA,Eisensmit RC,Woo SL. Detection of the Xmnl RFLP at the human PAH locus by PCR. Nucleic Acids Res. 1992; 20(4): 927.
4. Fang B, Yuan L, Wang M, Huang S, Wang T, Miao S, Ye J, Sun N, Lo H, Savio LC.Detection of point mutation of the phenylalanine hydroxylase gene and prenatal diagnosis of phenylketonuria. Chin Med Sci J. 1992; 7(4): 205-208.
5. 贺文萍,来暮,张乾忠,张新生,李秀玲. 中国两地区人群和日本人苯丙氨酸羟化酶基因突变差异的研究. 中华医学遗传学杂志1997; 14(6): 344-346.
6. 延正红,姚野崎,金慧敏,宋玉浩,宋晓良,成泽邦明. 16例苯丙酮尿症PAH基因Exon7突变的检测. 中华医学遗传学杂志1994; 11(5): 277-279.
7. Woo SL, Lidsky AS, Güttler F, Chandra T, Robson KJ. Cloned human phenylalanine hydroxylase gene allows prenatal diagnosis and carrier detection of classical phenylketonuria.Nature. 1983; 306: 151-155.
8. Levy HL. Molecular genetics of phenylketonuria and it sinoplication. Am J Hum Genet. 1989; 45: 667-670.
9. 李永旭. 《遗传代谢性疾病》pp123, 第一版, 人民出版社, 北京1982.
10. 袁丽芳,黄尚志,罗会元,方炳良,杨涛,王玫,王涛,孙念怙,赵时敏,刘慎如,叶珏. 16例经典型苯丙酮尿症的产前基因诊断. 中华医学遗传学杂志1995; 12(6): 327.
11. 郑梅玲, 化爱玲, 丁琰. 苯丙酮尿症高危胎儿的产前基因诊断. 中国优生与遗传杂志1999; 7(2): 20.
12. 黄尚志,方炳良,初海鹰,袁丽芳,王玫,许顺斌,罗会元. 苯丙氨酸羟化酶基因内短串联重复序列多态性的分析及应用. 中华医学杂志1995; 75(1):22-24.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




