JMC:新型STAT3抑制剂HP590,口服对胃癌有效
2022-09-23 精准药物 精准药物

信号传导及转录激活蛋白3(STAT3)是一类重要的转录因子,其常被发现在多种癌症中异常激活,参与肿瘤发生发展的多个过程,并与多种癌症的不良预后密切相关。因此,在过去二十年,STAT3作为潜在的抗肿瘤靶
2022年9月14日,国际药物化学权威期刊《Journal of Medicinal Chemistry》在线发表了华东师范大学陈益华/易正芳研究员团队合作的最新研究成果“Discovery of a Novel Potent STAT3 Inhibitor HP590 with Dual p-Tyr705/Ser727 Inhibitory Activity for Gastric Cancer Treatment”。这是继4月27日陈益华团队在同期刊发表“Discovery of 2-Amino-3-cyanothiophene Derivatives as Potent STAT3 Inhibitors for the Treatment of Osteosarcoma Growth and Metastasis”后在STAT3抑制剂研究方面取得的又一次重要突破。

图1.文章标题
近年来,靶向转录调控因子已成为开发新型抗肿瘤药物的热门研究领域之一。信号传导及转录激活蛋白3(STAT3)是一类重要的转录因子,其常被发现在多种癌症中异常激活,参与肿瘤发生发展的多个过程,并与多种癌症的不良预后密切相关。因此,在过去二十年,STAT3作为潜在的抗肿瘤靶点受到大量药物研究科学家和制药公司的青睐。目前,已有少量直接STAT3抑制剂进入临床用于不同癌症治疗的研究,但这些抑制剂仍存在体内抗肿瘤效果不足、成药性较差以及安全性不够等问题,更重要的是,大部分报道的STAT3抑制剂只能抑制STAT3的Tyr705位点磷酸化,而不能抑制STAT3另一个氨基酸残基Ser727的磷酸化。大量研究表明,STAT3的激活主要涉及两个氨基酸残基Tyr705和Ser727的磷酸化,两者在肿瘤细胞中发挥不同的生物学功能,Tyr705磷酸化主要介导STAT3的核转录功能,而Ser727磷酸化主要介导STAT3的线粒体氧化磷酸化功能,两者在多种肿瘤中高表达且均能促进肿瘤的恶性增殖或导致其他肿瘤发展过程。鉴于STAT3 Ser727磷酸化在多种肿瘤中同样发挥着重要作用,陈益华研究员团队与其合作者易正芳研究员团队致力于开发能同时抑制Tyr705和Ser727磷酸化的新型STAT3抑制剂。

图2.STAT3双磷酸化抑制剂的筛选策略及苗头化合物1a的发现
本研究中,作者们以STAT3双位点磷酸化在胃癌细胞发生发展的重要角色为背景,基于STAT3双位点磷酸化在肿瘤细胞中介导不同生物学功能建立了全新的STAT3双磷酸化抑制剂的高通量筛选实验,即表征STAT3 p-Tyr705抑制活性的STAT3依赖性荧光素酶实验(Luciferase reporter assay)和用于表征p-Ser727抑制活性的ATP抑制实验 (ATP inhibition assay)。随后,通过筛选大量化合物,作者获得一个能在微摩尔水平显著抑制STAT3荧光素酶活性、胃癌细胞中ATP生成以及多株胃癌细胞增殖的理想苗头化合物1a(图2)。为了进一步提高苗头化合物1a的生物活性,作者们进行了理性的药物化学设计与构效该系研究,最终导致了具有低纳摩尔抗肿瘤活性的先导化合物HP590的发现(图3)。同时,作者们还在本文中总结了这类三芳香杂环STAT3双磷酸化抑制剂的构效关系,其具体内容如图4所示。
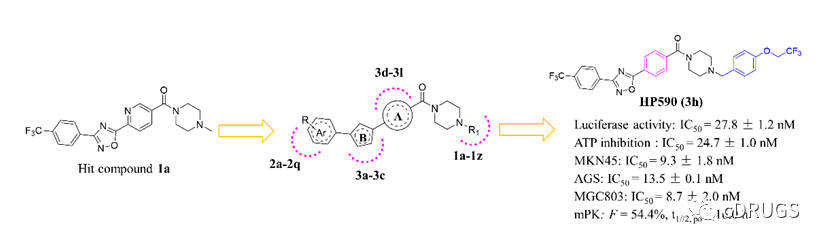
图3.从苗头化合物到先导化合物的结构优化过程
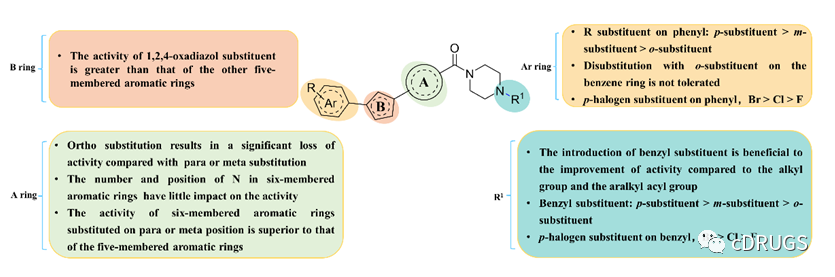
图4.改造化合物的构效关系总结
接下来的工作中,作者们对先导化合物HP590进行了系统的靶点验证和体内外抗肿瘤活性评价。首先,作者们在WB实验中验证先导化合物HP590是否能在肿瘤细胞中抑制STAT3的双位点磷酸化。结果显示,HP590能在低纳摩尔浓度下以时间梯度和浓度梯度依赖的方式抑制多株胃癌细胞中的STAT3双位点磷酸化,而对STAT3蛋白本身没有显著影响。随后,作者们通过分子对接(docking)、微量热泳动仪(MST)、细胞热转移实验(CETSA)实验证明HP590能以高亲和性和选择性的结合到STAT3的SH2结构域,而对与其高度同源的STAT1和STAT5无结合作用。另外,作者们还发现HP590对STAT3上游酪氨酸激酶和丝氨酸激酶无明显的抑制作用。这些证据表明HP590是一个直接的STAT3抑制剂,其可以通过靶向STAT3的SH2结构域显著抑制其双位点磷酸化(图5)。紧接着,作者们还通过调查HP590对胃癌细胞中STAT3下游、STAT3转入细胞核以及线粒体氧化磷酸化的抑制效果,结果显示HP590能同时抑制p-Tyr705和p-Ser727分别介导的核转录功能和线粒体氧化磷酸化功能,再次表明HP590是一个STAT3双位点磷酸化抑制剂。此外,作者还对HP590的药代动力学性质进行初步的评价,结果显示HP590具有较优的口服生物利用度以及体内代谢稳定性,同时对多种CYP450酶无明显抑制作用。
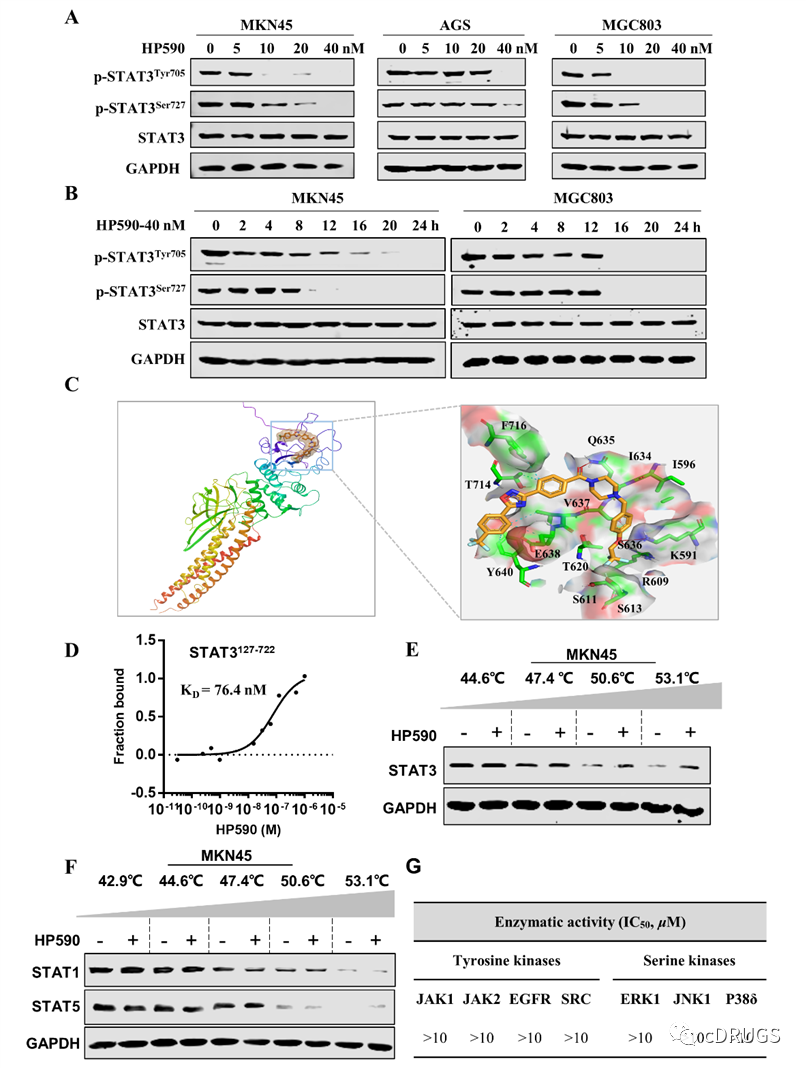
图5.HP590通过选择性靶向STAT3抑制其双位点磷酸化
最后,作者们对HP590的体内外抗胃癌细胞增殖活性进行评价。结果表明,HP590能选择性抑制胃癌细胞的增殖而对低表达STAT3双磷酸化的正常细胞无明显抑制,另外,HP590相比于阳性化合物BBI608能更加显著的抑制多株胃癌细胞的克隆形成以及诱导胃癌细胞的凋亡(图6)。同时,作者们还发现HP590对敲低STAT3的胃癌细胞无明显抑制,再次表明STAT3是HP590在体外发挥抗肿瘤活性的主要靶点(图6)。体内研究表明,口服HP590显著抑制MKN45异种模型的增殖,而且通过对HP590治疗后的肿瘤组织分析发现,HP590在体内同样显著抑制STAT3双位点磷酸化和下游基因的表达,表明STAT3是HP590在体内发挥抗肿瘤活性的主要靶点(图7)。重要的是,HP590在治疗过程中对小鼠体重和主要器官无明显影响,表明HP590是一类安全有效的STAT3双磷酸化抑制剂,可为未来胃癌的治疗提供新的可能。
总结:该研究报道了一种全新的STAT3双磷酸化抑制剂的高通量筛选策略,阐述了从苗头化合物的发现到先导化合物改造和构效关系研究过程,通过靶点的验证以及体内外的抗肿瘤活性研究确定HP590可作为一类新型的选择性STAT3双位点磷酸化抑制剂,为下一代STAT3抑制剂开发提供新的思路。
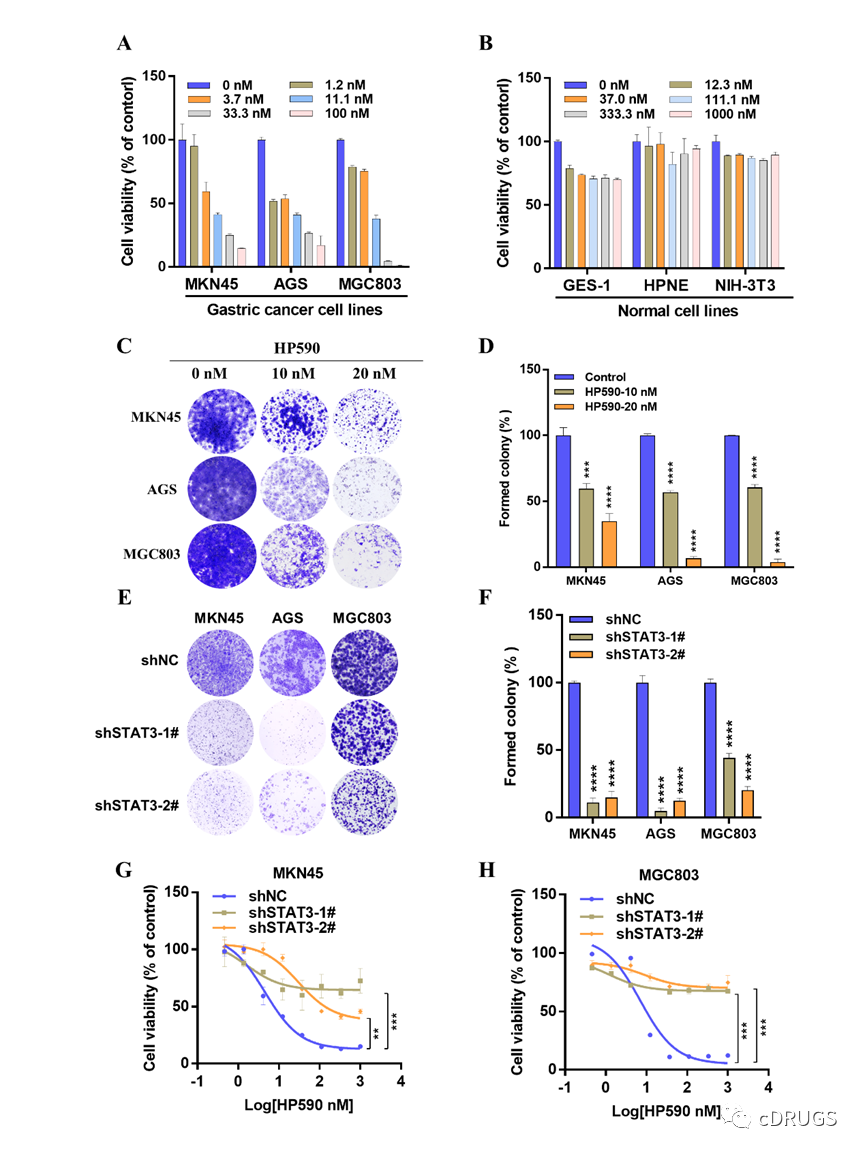
图6.HP590在体外显著抑制胃癌细胞的增殖
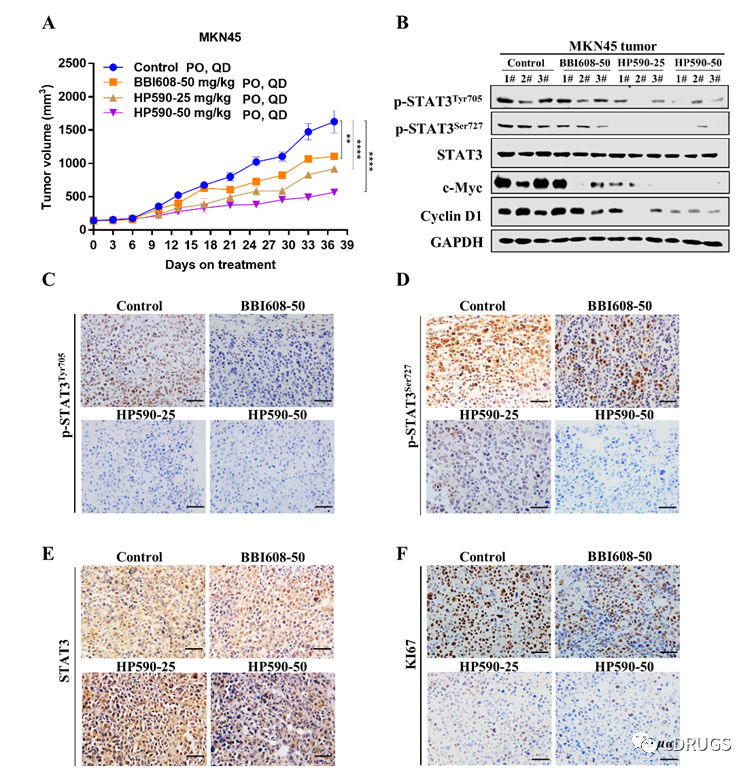
图7.HP590在体内通过抑制STAT3双位点磷酸化显著抑制胃癌细胞的增殖
该文章以华东师范大学陈益华研究员和易正芳研究员为共同通讯作者,华东师范大学博士生何朋和博士生缪颖为共同第一作者,另外,该研究获得华东师范大学刘明耀教授的大力支持。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00413

“精准药物”公众号由暨南大学药学院张章博士创建。张章博士主要从事:1)小分子靶向抗肿瘤药物的发现、作用机制和成药性研究;2)基于化学蛋白质组学的疾病和药物靶标发现研究。作为主要完成人,团队开发的4个激酶抑制剂先后转让给企业进行后期开发,其中1个已上市(奥雷巴替尼),1个报生产,1个在I期临床,1个在临床前研究阶段。实验室具备完善的体内外药效、机制、成药性、靶标发现和确证等技术平台;支撑和推动了多个企业靶向抗肿瘤药物的发现、成药性评价、新适应症和机制等研究。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言