先天性肌无力综合征:症状与体征、病因、流行病学、诊断与治疗
2022-09-16 MedSci原创 MedSci原创

先天性肌无力综合征(congenital myasthenic syndrome,CMS) 是以疲劳性肌无力为特征的一组遗传性疾病。由于神经肌肉接头的突触前、突触基膜和突触后结构的遗传缺陷导致运动终板
先天性肌无力综合征(congenital myasthenic syndrome,CMS) 是以疲劳性肌无力为特征的一组遗传性疾病。由于神经肌肉接头的突触前、突触基膜和突触后结构的遗传缺陷导致运动终板神经肌肉接头信息传递受损。CMS 好发于青少年、儿童和婴幼儿。主要临床特征包括四肢近端无力、延髓麻痹、呼吸衰竭。根据 CMS 病变部位分为突触前膜、突触间隙、突触后膜病变、糖基化缺陷和肌病重叠综合征。
一、一般概述
CMS涉及神经肌肉接头,这是一个突触,运动神经的信号在这里传递给肌肉纤维,并告诉肌肉纤维何时收缩。
正常的神经肌肉接头由突触前区域、突触空间和突触后区域组成。突触前区包含运动神经细胞的末端,称为运动神经末梢。运动神经末梢覆盖着肌肉纤维的一个特殊区域,称为突触后区域。运动神经末梢和突触后区域之间的空间被称为突触空间或突触裂隙。突触后区域显示多个褶皱,被称为交界褶皱。运动神经末梢含有小囊泡,由神经递质乙酰胆碱(简称ACh)填充,作为化学 "信使",为肌肉收缩提供指令。
覆盖运动神经终端并面向突触空间的膜被称为突触前膜。覆盖突触后区域的膜被称为突触后膜。突触后膜覆盖结节褶皱尖端的部分由乙酰胆碱受体分子衬托,简称AChR。突触空间由被称为突触基底膜的膜衬托。这层膜固定着乙酰胆碱酯酶的分子,简称AChE,这是一种将乙酰胆碱转化为醋酸和胆碱的酶。
运动神经末梢与肌肉纤维的交流过程是一个高度专业化的过程,损害这种交流的遗传缺陷会导致先天性肌萎缩综合症。了解这一过程有助于理解肌萎缩性疾病。
当肌肉处于静止状态时,运动神经终端的单个突触囊泡会随机释放乙酰胆碱。这种释放被称为 "细胞外渗"。从单个突触小泡中释放的乙酰胆碱的数量被称为乙酰胆碱的量子。
从突触小泡中释放的乙酰胆碱通过突触空间,与集中在连接褶皱顶端的乙酰胆碱结合。当这种结合发生时,它导致AChR中心的一个通道,并允许带正电的钠离子和少量的钙离子进入肌肉纤维。这一过程短暂地将突触后膜上的电荷从负极变为正极(突触后的小去极化),这被称为微型内板电位(MEPP)。
当一个人想做一个自愿的动作时,(如举手、跳舞、踢球等),一系列连续的神经冲动被送到运动神经末梢,它们使突触前膜去极化,导致称为电压门控钙通道的结构打开,使钙进入运动神经末梢。这种钙的涌入导致几个突触小泡的内容物几乎同步释放,从而使突触后膜出现更大的去极化,这被称为板内电位(EPP)。当EP达到一定的阈值时,它打开沿运动终板区域外的整个肌肉纤维发现的电压门控钠离子通道,这引发了传播的肌肉纤维动作电位,导致肌肉纤维收缩。
内板电位与激活电压门控钠离子通道所需的去极化之间的差异被称为神经肌肉传导的安全系数。在健康人中,EPP的振幅是相当大的。随着持续的活动,EPP开始下降,但仍然大到足以触发肌肉纤维动作电位。
肌肉收缩后,ACh从AChRs释放到突触空间,在那里它被AChE分解(水解)为两个分子,即乙酸和胆碱。胆碱被运回神经末梢,在一种被称为胆碱乙酰转移酶的影响下,它与乙酸盐重新结合,再次储存在突触小泡中。
管理神经肌肉传递安全系数的因素主要分为四类:(1)影响突触小泡中ACh分子数量的因素;(2)影响定量释放机制的因素;(3)突触空间中AChE的密度;以及(4)影响单个量子效能的因素。单个量子的功效取决于内板的几何形状、AChRs在连接褶皱顶端的包装密度、AChRs对ACh的亲和力以及AChR离子通道的动力学特性。
先天性肌无力综合征是由一个特定基因的改变(突变)引起的。这导致了一种异常的蛋白质或甚至是一种蛋白质的丢失,损害了上述过程的某些部分。异常蛋白(疾病蛋白)可以驻留在运动神经末梢,或突触空间,或位于神经末梢下的突触后区域,但在一些患者中,疾病蛋白也存在于其他组织或器官中,不仅引起CMS,而且还引起各种其他症状。
二、症状与体征
所有肌无力症的主要症状是因劳累而引起或加重的肌肉无力。这被称为疲劳性肌无力。在健康人中,体力活动会使神经末梢释放的ACh量子数量略有减少,但这并不影响神经肌肉传导的安全系数,但对于肌无力患者来说,其安全系数已经降低。
在一些CMS患者中,肌无力局限于由颅神经供应(支配)的肌肉,导致双目失明、眼睑下垂(眼睑下垂)、面部无力、低鼻音或口齿不清以及吞咽困难。在其他病人中,上述症状与四肢和躯干肌肉的无力结合在一起,造成全身性肌无力症。还有一些患者的肌无力仅限于肢体和躯干肌肉,造成 "肢体-腰部肌无力症"。
由蛋白质糖基化所需的酶的缺陷引起的肌无力症还可能与发育迟缓、癫痫发作、智力障碍、神经病变和不同器官的代谢异常有关。
目前已发现几种不同类型的CMS1。
- 突触前
- 突触基底层相关的
- 乙酰胆碱受体的缺陷
- 正常内板发育和维持所需的蛋白质的缺陷
- 先天性的糖基化缺陷
- 其他肌无力症候群
每种类型都可以细分为几个亚型,下面将讨论。
(一)突触前cms
1、内板胆碱乙酰转移酶(ChAT)的缺陷
乙酰胆碱从神经末梢释放出来后,与乙酰胆碱受体短暂结合;当它从受体释放出来后,被乙酰胆碱酯酶迅速分解(水解)为胆碱和醋酸。释放的胆碱被运送到神经末梢,在那里胆碱乙酰转移酶(ChAT)对乙酰胆碱进行改造(重新合成)。重新合成的乙酰胆碱随后被运送到突触小泡中,在那里它可以根据需要被释放到突触空间。CHAT基因的可逆性突变,单独或结合在一起,改变了ChAT蛋白的表达、催化效率或稳定性。
ChAT的缺陷导致活动期间突触小泡中的乙酰胆碱含量逐渐减少,从而降低了EPP的振幅,降低了神经肌肉传导的安全系数。
一些病人在出生时表现为肌张力低下(肌张力低)、颅骨和四肢肌肉瘫痪和呼吸暂停(不能呼吸)。有些病人出生时是正常的,在婴儿期或儿童期因感染、兴奋或无明显原因而发作呼吸暂停,3-7有些儿童急性发作后出现持续数周的呼吸衰竭。其他患者随着年龄的增长而改善,但仍有不同程度的眼睑下垂(上睑下垂),眼部肌肉运动障碍,易疲劳的虚弱,以及反复发作的发绀,即由于呼吸障碍和血液含氧量不足导致的皮肤发蓝。有些病人只抱怨有轻度至中度的疲劳性虚弱。暴露在寒冷的环境中会使虚弱加剧,因为这进一步降低了变异酶的效率。
治疗包括吡啶斯的明(Mestinon)的预防(预防性)治疗,这是一种抑制乙酰胆碱酯酶(在突触空间分解乙酰胆碱)活性的药物。这延长了突触空间中乙酰胆碱的寿命,从而延长了它能激活的乙酰胆碱受体的数量。应向受影响婴儿的父母提供一个充气式救援袋和一个合适的面罩,并指导他们肌肉注射甲基硫酸新斯的明(另一种乙酰胆碱酯酶的抑制剂),并建议他们在家中安装一个呼吸监测器。
2、SNAP25B肌萎缩症
迄今为止,报告的一名患者患有严重的CMS,伴有对大脑刺激异常夸张的反应(大脑皮层过度兴奋)、共济失调(缺乏协调性)和智力障碍。9 遗传学研究发现SNAP25B基因中存在一个显性的单氨基酸变化,该基因产生一种突触小泡释放乙酰胆碱(外渗)所需的基本蛋白。用电子显微镜检查时,内板结构正常。用3,4-二氨基吡啶(3,4-DAP)治疗,可增加神经冲动所释放的量子的数量,改善了病人的虚弱,但没有改善她的共济失调或智力障碍。
3、突触凝集素2缺乏症
突触凝集素2(Synaptotagmin 2)是另一种突触前蛋白。它感知神经末梢的钙浓度;当钙浓度增加时,它作用于其他蛋白,启动乙酰胆碱释放到突触空间(外渗)。在两个亲缘关系中,该基因的突变导致肢体肌肉无力,腱反射消失,以及肌肉纤维收缩前的肌肉纤维动作电位的振幅降低。这种反应以及无力的情况通过运动得到短暂的改善。对这种情况的治疗还没有描述。
4、突触小泡的缺乏和定量释放的障碍(Paucity of Synaptic Vesicles and Impaired Quantal Release)
其临床特征与自身免疫性肌无力相似,但在出生时或婴儿早期发病,抗乙酰胆碱受体抗体的检测为阴性。具体的诊断需要对运动终板进行电子显微镜和电生理学研究。突触前缺陷表现为神经冲动所释放的乙酰胆碱数量严重减少(约为正常的20%),同时神经末梢的突触小泡数量也相应减少。这种CMS对吡啶斯的明的治疗有反应。
(二)突触基底层相关的
1、内板乙酰胆碱酯酶(AChE)的缺乏
内板种乙酰胆碱酯酶(AChE)由12个催化亚单位组成,可迅速分解(水解)乙酰胆碱,加上一个胶原亚单位,称为ColQ,将整个分子固定在内板的基底板上。亚单位是单一的蛋白质分子,与其他蛋白质结合形成一个更大的蛋白质复合物。
ColQ蛋白由三条相同的链组成,每条链与4个催化亚单位结合。组织化学和电子显微镜研究显示,内板没有乙酰胆碱酯酶,神经末梢比正常小。严重的患者在出生时出现呼吸暂停和全身无力,并持续一生。12 这些病人对抑制乙酰胆碱酯酶的吡啶斯的明没有反应,或病情加重。治疗仍不理想,但麻黄素13和阿布特罗14有逐渐发展的有益作用。
2、与β2-Laminin缺乏有关的CMS(CMS Associated with β2-Laminin Deficiency)
β2-层粘连蛋白是不同组织基底层的组成部分,在肾脏、眼睛和内板上高度表达,在内板上该蛋白对神经末梢与突触后区域的适当排列很重要。β2-层析蛋白还有助于这两个区域的发育和组织。β2-层粘连蛋白的突变导致了皮尔森综合征,这是一种罕见的疾病,与肾脏和眼睛的畸形有关。一个患有Pierson综合征的病人有肌萎缩综合征。肾脏缺陷在15个月大时通过肾脏移植得到了纠正。神经冲动的定量释放和MEPP的振幅都减少了。电子显微镜显示神经末梢异常地小,这就是定量释放减少的原因。突触空间变宽,结节褶皱简化,这是MEPP振幅下降的原因。
(三)乙酰胆碱受体(AChR)的缺陷
1、原发性乙酰胆碱缺乏症(Primary AChR Deficiency)
乙酰胆碱受体是由5个亚单位组成。亚单位是单一的蛋白质分子,与其他蛋白质结合形成一个较大的蛋白质复合物;在这种情况下,就是乙酰胆碱受体。其中两个亚单位被称为α(α),其余三个亚单位在成人中被称为β(β)、δ(δ)和ε(ε)。在出生前,胎儿的亚单位包含一个γ(γ)而不是ε亚单位。
在原发性AChR缺陷中,在终板表达的AChR数量减少,神经肌肉传导的安全系数因EPP的振幅降低而受损。临床缺陷从轻微到严重不等。ε亚单位隐性突变的病人一般比其他亚单位突变的病人病情轻,因为胎儿γ亚单位的代偿性表达可以部分替代有缺陷的ε亚单位。
病情最严重的病人从出生起就有严重的眼肌、球状肌和呼吸肌无力,只能靠呼吸支持和灌食来生存。灌胃式喂养是指用一根小而窄的管子从婴儿的鼻孔插入,顺着喉咙流向胃部,直接向患儿提供营养。婴儿在出生后的第一年可以脱离呼吸机并开始耐受口服喂养,但在儿童期和成年后会出现吸入性肺炎,可能需要间歇性的呼吸支持。
运动发育严重延迟;他们很少学会迈步,只能走一小段路。老年患者通过用手支撑下颌来闭口,用手指抬高眼睑。面部畸形、下巴突出、牙齿错位(错牙合)和脊柱的异常弯曲,如脊柱侧弯或脊椎侧弯,在第二个十年期间变得很明显。肌肉体积减少。腱反射正常或减弱。
不太严重的病人从儿童早期就有中等程度的身体障碍。眼球运动受限和眼睑下垂在生命的第一年变得明显。他们容易疲劳,走路和上楼梯都很困难,在运动中不能跟上同龄人,但可以进行大多数日常生活活动。减少或阻止AChR表达的AChRα、β和δ亚单位的突变在胚胎期是致命的,或在出生后引起明显的残疾和高死亡率。
在受影响最小的病人中,运动发育只是轻微延迟;他们只有轻微的眼睑下垂和眼球运动受限。他们在运动中往往很笨拙,容易疲劳,不能很好地跑步、爬绳或做俯卧撑。在一些病人中,当他们在外科手术中接触到神经肌肉阻断剂药物后出现长时间的呼吸停止时,才怀疑是肌无力症。
治疗包括吡啶斯的明,一种乙酰胆碱酯酶的抑制剂。这种药物增加了突触空间中乙酰胆碱的寿命,使每个乙酰胆碱分子在通过扩散离开突触空间之前能够反复与不同的乙酰胆碱受体结合。许多病人从使用3,4-二氨基吡啶(3,4-DAP)16中获得额外的好处,它延长了神经冲动对突触前膜的去极化。这使得更多的钙进入神经末梢,从而增加每个神经冲动所释放的乙酰胆碱量子的数量。最后,一些病人还能从阿布特罗中获得额外的好处。17
2、乙酰胆碱的动力学缺陷:慢通道综合征(Kinetic Defect in AChR: The Slow-Channel Syndrome)
这种综合征是由乙酰胆碱受体(AChR)基因的显性突变引起的,导致AChR离子通道的异常缓慢关闭。离子通道的长时间开放导致突触后区域带正电的离子(包括钙)过载。局部钙离子浓度的增加损害了连接处的褶皱,并可损害褶皱下的肌纤维核。症状的发生从婴儿期到成年早期不等。该病导致颈部、肩胛骨、腕部和手指伸肌选择性地严重无力和丧失体积(萎缩)。
突触传递的安全系数受到损害,原因是乙酰胆碱受体的丧失导致结节皱褶受损,以及受体在生理活动中因长期接触乙酰胆碱而变得不敏感(无反应)。
这种综合征对吡啶斯的明没有反应,或因吡啶斯的明而恶化,但会因相对大剂量的氟西汀(百忧解)而得到改善,氟西汀可以阻断(塞住)乙酰胆碱受体离子通道,从而减少通道开放的长度。
3、乙酰胆碱受体的动力学缺陷:快速通道综合征(Kinetic Defect in AChR: The Fast-Channel Syndrome)
这种综合征通过隐性遗传传播,在生理和解剖上与慢通道综合征相反。18 乙酰胆碱受体通道开口的长度减少,是因为突变降低了乙酰胆碱受体结合乙酰胆碱的能力,或阻碍了乙酰胆碱受体离子通道的开放,或导致离子通道变得间歇性不稳定。内板的结构完整性不受影响。
只有当AChR亚单位基因的第二个拷贝不表达,或基因的两个拷贝都怀有相同的突变时,这种综合征才会变得明显,因此,快速通道突变决定了临床后果。由于突变基因降低了通道开放的概率和长度,从而降低了EPP的振幅和持续时间,所以神经肌肉传导的安全系数下降。临床后果从轻微到严重不等。大多数病人对吡啶斯的明和3,4-DAP联合治疗有反应。
4、由AChR亚基和其他特定蛋白的突变引起的产前CMS
第一个被确认的产前肌无力综合征被追溯到胎儿AChR γ亚单位的突变。在人类中,怀有胎儿亚单位的AChR在妊娠第九周左右出现在发育中的肌肉纤维上,并在妊娠第十六周左右集中在早期神经-肌肉连接处。随后,γ亚单位被成年的ε亚单位取代,在妊娠31周后不再出现在胎儿的内板上。因此,γ亚单位的有害突变在妊娠第十六周至第三十一周期间减少了胎儿的运动(运动不足)。
出生时的临床后果是大关节挛缩,肌肉体积小,颈部、腋下、肘部、手指或膝盖后面有蹼,手指屈曲挛缩,脚跟突出的摇椅底,以及具有轻度眼睑下垂和嘴角下垂的典型面部外观。挛缩是指关节永久地固定在弯曲或伸直的位置,完全或部分地限制了受影响关节的活动。
最近的研究还发现,由于AChR α、β和δ亚基的两个拷贝以及其他CMS疾病基因的有害空突变,导致了致命的胎儿运动障碍综合征。
(四)由内板发育或维持缺陷引起的cms
迄今为止,在对运动终板的发育和维持至关重要的蛋白质的基因中已经检测到突变。如同上述从神经细胞到肌纤维的神经信号交流一样,运动终板的健康和发育取决于一系列相互关联的化学反应,涉及多个基因及其蛋白产物。
这些基因是MuSK、Agrin、LRP4和DOK-7。Agrin由神经终端分泌到突触空间,在那里它与突触后膜上的脂蛋白相关蛋白LRP4结合,形成Agrin-LRP4蛋白复合物。然后Agrin-LRP4复合物与MuSK结合并激活MuSK。这增强了MuSK的磷酸化,导致LRP4和MuSK的聚集。激活的MuSK与突触后的DOK-7和其他突触后的蛋白一起作用于Rapsyn,使AChR集中在突触后的膜上,增强突触后核的特定基因表达,并促进突触后分化。聚集的LRP4又能促进运动轴突的分化。Agrin-LRP4-MuSK-Dok-7信号系统对维持成人神经肌肉接头的结构也很重要。
1、Agrin缺乏症
目前仅有少数与粒蛋白相关的CMS患者被报道过。症状的严重程度因agrin基因突变的位置和突变是否影响agrin的表达而不同。23,24,25当阻碍agrin附着于LRP4的突变在临床上占主导地位时,后果就很严重。在这样的病人中,突触接触分散,突触后区域分化差,神经末梢小,结节褶皱下的肌纤维有退行性改变。24 另一份报告描述了三个亲属关系,其中agrin突变与缓慢进行的腿部远端和后来的上臂肌肉萎缩有关。25 agrin相关CMS的治疗并不令人满意,但一个病人对麻黄碱有部分反应。
2、LRP4缺乏症
只有两份关于LRP4相关的CMS的报告。第一份报告描述了一个17岁的女孩,有中度严重的疲劳性肢体无力,突触接触形状不规则,有轻度内板AChR缺乏。在该患者位于肋骨之间的肌肉(肋间肌)中,MEPPs和EPPs的振幅正常,表明所发现的突变可使某些肌肉的神经肌肉传导得以保留。26 随后,有两姐妹患有中度严重的CMS,并携带一个阻碍LRP4激活MuSK的同源突变,被证明具有结构和功能异常的内板和内板AChR缺乏症。
3、MuSK缺乏症
这种疾病在出生时或生命早期表现为眼睑下垂或呼吸困难。28,29,30,31,32 在小鼠中引入突变基因会导致神经供应的局部丧失(去神经化)和神经供应的重建(再神经化)的反复循环,导致内板的广泛重塑。最近的一份报告指出,在两兄弟中使用阿布特罗尔治疗非常有效。
4、DOK-7缺乏症
DOK-7在发育和成熟的肌肉纤维中表达。在发育中的肌纤维中,它作为MuSK的内在激活剂。35在成熟的肌肉中,它被MuSK激活,以激活rapsyn,使乙酰胆碱受体集中在交界处的褶皱上,并促进内板的发育和维持。这种CMS可以是温和的,也可以是严重的。致病性突变可遏制DOK-7的表达,或阻止DOK-7激活或被其他细胞内蛋白激活。确定的突变与临床特征之间似乎没有一致的关联。
所有受影响的病人都有四肢无力,面部和颈部肌肉无力较轻,但少数病人有严重的球部无力,少数病人的眼球运动明显受限。椎间盘的维持功能受损,表现为椎间盘的持续破坏和重塑。重要的是,这种CMS在使用吡啶斯的明后会迅速恶化,但在一段时间内对麻黄素38或阿布特罗的反应良好
5、Rapsyn缺乏症
Rapsyn使乙酰胆碱受体集中并固定在结节皱褶上39,是结节皱褶发育所必需的。41 出生时的关节挛缩和其他先天性畸形的发生率接近三分之一。42 同时发生的感染或发热可引发呼吸危机,由于缺氧(缺氧)可导致脑损伤。43,44 大多数病人的眼球运动是完整的。42 单个肌纤维上出现多个突触接触。内板乙酰胆碱受体缺乏症比原发性乙酰胆碱受体缺乏症42要轻一些,而且交界处的褶皱分化不明显。大多数病人对吡啶斯的明反应良好;有些人从麻黄素或阿布特罗43中获得额外的好处,有些人通过3,4-DAP进一步改善。
印欧人在产生rapsyn的基因中存在一种常见的N88K突变,这涉及到在密码子8845处用赖氨酸分子(K)替换天冬酰胺分子(N)(密码子:3个相邻核苷酸的序列,构成一个特定氨基酸的遗传密码)。不同的突变会阻碍rapsyn分子的自我联合,或与乙酰胆碱受体的结合,或阻碍agrin-MuSK-LRP4介导的这些受体的聚集,或减少rapsyn的表达。没有基因型与表型的相关性(某一突变与临床特征之间的相关性),但具有同型E-box突变(E-box:在参与调节基因表达的基因编码区之前的序列)的近东犹太患者的病程较轻,眼睑下垂,下颌大突起,咀嚼肌和面肌严重无力,说话时鼻音过重47。
(五)与先天性糖基化缺陷有关的肌萎缩综合征
糖基化是糖 "树 "或残基(糖类)被创造、改变和化学地连接到某些蛋白质或脂肪(脂类)的过程。当这些糖分子与蛋白质相连时,它们形成糖蛋白;当它们与脂质相连时,它们形成糖脂。糖蛋白和糖脂在所有组织和器官中具有许多重要功能。糖基化涉及许多不同的基因,编码许多不同的蛋白质,如酶。这些酶之一的缺陷或缺乏可导致各种症状,可能影响多个器官系统。
对新生的肽进行糖基化可以增加它们的溶解度、折叠、稳定性、组装和细胞内运输。肽是氨基酸化合物,可以在体内发挥广泛的功能。O-糖基化涉及在氨基酸丝氨酸和苏氨酸上添加糖残基;N-糖基化发生在连续的步骤中,对氨基酸天冬酰胺的氨基进行装饰。
到目前为止,有四种负责N-糖基化的酶的缺陷已被证明可导致CMS。GFPT1,50,51 DPAGT1,52,53 ALG2和ALG14.54 肌肉纤维内的小管状物堆积,被称为小管状物,是诊断的线索,但并非所有患者都能看到。由于糖基化蛋白存在于所有的终板部位,神经肌肉传导的安全系数很可能被突触前和突触后的缺陷共同影响。
1、GFPT1缺陷
GFPT1控制葡萄糖进入糖基化途径。GFPT1的缺陷预示着糖基化的减少,因此也预示着几个内板相关蛋白的功能有缺陷。她有空泡性肌病,神经冲动诱发的定量释放减少,而且MEPP振幅低。空泡性肌病是一种肌肉疾病,与肌肉组织内被称为空泡的不正常口袋或空间的发展有关。
2、DPAGT1缺乏症
DPAGT1催化N-链接蛋白质糖基化的第一个承诺步骤。DPAGT1缺陷预示着分布在整个机体内的多种蛋白质的天冬酰胺糖基化受损,但在首批5名携带DPAGT1基因突变的患者中,只有神经肌肉传导受到不利影响。52随后对两个兄弟姐妹和第三个患者的研究显示,DPAGT1缺陷与智力障碍有关。9兄弟姐妹对吡啶斯的明和3,4-DAP反应不佳;第三个患者通过吡啶斯的明和阿布唑部分改善。肋间肌研究显示纤维类型不相称,一些肌纤维中有小管状聚集物,以及自噬性空泡(降解和消化亚细胞结构的空泡)。诱发的定量释放、MEPP振幅和内板乙酰胆碱受体含量都减少到正常的50%。
3、ALG2和ALG14的缺失
ALG2催化N-糖基化的第二和第三步骤。在一个家族中,四个受影响的兄弟姐妹有一个有害的同型突变,第三个病人是同型的低表达突变。ALG14与ALG13和DPAGT1形成一个酶复合物,也有助于N-糖基化的第一个承诺步骤。在一个家庭中,两个受影响的兄弟姐妹携带了两个不同的隐性突变。内板超微结构和神经肌肉传导的参数未被调查。
(六)其他先天性肌萎缩综合征
1、PREPL缺失综合征
肌张力低下-胱氨酸尿症综合征是由染色体2p21的SLC3A1和PREPL基因的隐性缺失引起。主要的临床特征是胱氨酸尿症、生长激素缺乏、肌肉无力、眼睑下垂和喂养问题。胱氨酸尿症是一种遗传性代谢疾病,其特点是某些有机化合物(氨基酸)在肠道和肾脏的异常移动(运输)。
一个孤立的PREPL缺乏的患者自出生以来就有肌萎缩症状和生长激素缺乏,但没有胱氨酸尿症,并在婴儿期对吡啶斯的明有短暂的反应。55她的PREPL基因有一个父系遗传的无义突变和一个母系遗传的涉及PREPL和SLC3A1的缺失,因此PREPL缺乏决定了表型。患者的肌肉和内板中没有PREPL的表达。内板研究显示,尽管内板乙酰胆碱受体表达旺盛,但诱发的定量释放减少,MEPP振幅小。55由于PREPL是凝集素相关适配蛋白1(AP1)56的基本激活剂,而AP1是囊状乙酰胆碱转运体所需要的,以使突触小泡充满乙酰胆碱57,小MEPP被归因于囊状乙酰胆碱含量减少。
2、Na-通道肌无力症
迄今为止,已发现两名患有这种综合征的病人。第一位患者自出生以来就有突然发作的呼吸和面部无力,并伴有说话和吞咽所需的肌肉无力,持续3至30分钟,是典型的周期性麻痹以及肌无力症。对神经肌肉传导的研究显示,正常振幅的EPPs经常不能产生肌肉动作电位,指出电压门控钠通道(SCN4A基因)是罪魁祸首。电压门控钠离子通道(SCN4A)的基因有两个隐性突变,导致钠离子通道在被EPP激活后很快失去活性。在该患者中,电压门控钠通道的两个不同的隐性突变导致钠(Na)通道因活动而异常失活。
3、由Plectin缺失引起的CMS
由PLEC基因编码的Plectin有不同的组织特异性和细胞器特异性形式(称为异构体),其作用是将细胞骨架丝与目标细胞器连接起来,60-62细胞器是活细胞内任何数量的有组织或专门结构的总称。
Plectin集中在机械压力的部位。例如,在骨骼肌中,它存在于内板的连接褶皱下、肌肉纤维的表面膜下、Z-盘(标记相邻收缩单元边界的薄蛋白带),以及细胞核和线粒体周围,这些细胞几乎在身体的每个细胞中都有数百个,并产生大部分的细胞能量。
在皮肤中,它与半透明体(连接上皮细胞和底层基底膜的钉状结构)有关。单独或结合使用,plectin的突变可导致一种被称为单纯性表皮松解症(EBS)的水疱性皮肤病,也可导致进行性肌营养不良症,有时还可导致肌萎缩综合征。笔者调查的两名病人患有EBS,这是一种肌萎缩综合征,是由于结节褶皱退化引起的低振幅MEPPs,以及与肌纤维核、线粒体和肌纤维(肌肉细胞的基本杆状单位)脱位有关的肌萎缩,以及肌纤维表面膜的缺陷导致钙超载和肌纤维变性。
4、与线粒体柠檬酸合成酶载体SLC25A1缺陷有关的CMS
SLC25A1基因编码一种转运蛋白,负责柠檬酸盐在线粒体内膜的移动。SLC25A1基因的突变会干扰大脑、眼睛和精神运动的发育。
父母为近亲的两个兄弟姐妹有一个与智力障碍有关的CMS,全外显子组测序显示他们携带了SLC25A1的同源突变。随后的研究表明,该突变损害了该酶的运输活性,在斑马鱼中相当于SLC25A1的基因被敲除后,阻碍了运动轴突对肌肉纤维的支配。65 第三位患者携带SLC25A1的两个隐性突变,有肌萎缩症状,也有视神经发育不全、胼胝体(连接两个大脑半球的结构)不发育,以及尿中排泄过多2-羟基戊二酸。
5、与核中心型肌病有关的CMS
眼睑下垂、外眼角和面部肌肉无力、运动不耐受、肌电图研究下降以及对吡啶斯的明的反应,已被记录在由羊角蛋白(BIN1)66、肌管蛋白(MTM1)67和达那敏2(DNM2)68的突变引起的核心肌病(CNM)患者以及其他没有发现突变的CNM患者中。
三、病因
先天性肌萎缩综合征是由特定基因的改变(突变)引起的。基因提供了创造蛋白质的指令,这些蛋白质在身体的许多功能中发挥着关键作用。当一个基因发生突变时,蛋白质产品可能有问题,效率低下,或没有。根据特定蛋白质的功能,这可能影响身体的许多器官系统。
目前已知约有30个不同的基因会导致CMS。这些基因含有对神经肌肉接头和运动终板的正常功能或健康至关重要的蛋白质的指令。其中一些蛋白质存在于身体的其他部位,在这些亚型中,除了神经肌肉接头外,身体的其他部位也会受到影响。
在一些患有CMS的个体中,没有发现改变的基因,这表明存在着额外的、尚未确定的基因,可以引起先天性肌萎缩综合征。
在大多数亚型中,CMS是作为常染色体隐性遗传的。遗传病是由父亲和母亲的染色体上的特定性状的基因组合决定的。当一个人从父母双方都继承了同一性状的同一异常基因时,就会发生隐性遗传病。如果一个人接受了一个正常的基因和一个疾病的基因,这个人将是疾病的携带者,但通常不会出现症状。两名携带者的父母同时传递缺陷基因的风险是25%,因此,每次怀孕都会有一个受影响的孩子。每次怀孕时,生出像父母一样的携带者的孩子的风险是50%。一个孩子从父母双方获得正常基因并在该特定性状方面遗传正常的机会是25%。男性和女性的风险是一样的。
特定的CMS亚型,特别是SNAP25、synaptotagmin 2和慢通道-肌萎缩综合征是通过常染色体显性遗传来传播的。显性遗传病发生时,只有一个异常基因的单拷贝才会导致某种疾病。异常基因可以从父母任何一方遗传,也可以是受影响个体的新突变(基因改变)的结果。每次怀孕时,异常基因从受影响的父母传给后代的风险是50%。这种风险对男性和女性来说是一样的。
四、流行病学
国外报道 CMS 的平均发生率约每 100 万青少年儿童中 9.2 人。我国尚无该病流行病学数据。CMS 绝大部分是常染色体隐性遗传,有家族史的少见,一些常染色体显性遗传的 CMS 综合征可有家族史。
五、鉴别诊断
CMS 的鉴别诊断主要是需要与先天性肌病、肌营养不良、重症肌无力(MG)、兰伯特 - 伊顿综合征(Lambert-Eaton syndrome,LES) 相鉴别。主要鉴别点如下:
1. 发病年龄 CMS 绝大部分于出生后或婴幼儿起病,也有少数起病较晚者。先天性肌病、肌营养不良好发于婴幼儿,MG 和 LES 好发于成年期。
2. 症状与体征 CMS 表现为波动性肌无力、疲劳不耐受,而先天性肌病、肌营养不良多为持续性肌无力。CMS 多于颅面部肌无力起病,先天性肌病早期影响四肢近端,重症肌无力和 Lambert-Eaton 综合征早期表现为眼外肌麻痹、四肢近端易疲劳。
3. 电生理 R-CAMP 现象和运动后重频递减提示 CMS 可能,但非必要条件。CMS、MG 和 LES 可出现重复电刺激低频递减,Jitter 阻滞和增宽表现,上述现象一般不出现在先天性肌病和肌营养不良症中。
4. 全身表现 少数 CMS 可出现关节挛缩、脊柱侧弯,与某些先天性肌病和肌营养不良相似,但不出现在 MG 和 LES 中。
5. 抗体 CMS 外周血的乙酰胆碱受体抗体和 MuSK 抗体为阴性,大多数 MG 中上述抗体为阳性。
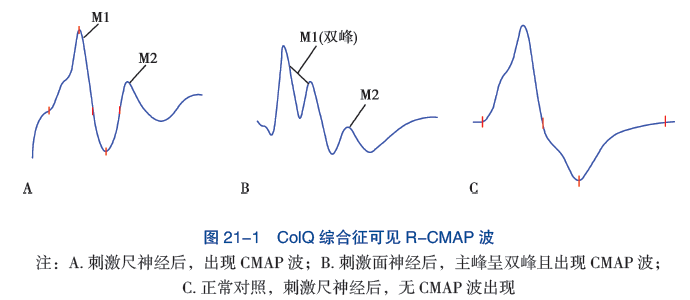
六、诊断
临床上可以根据自婴儿期或幼儿期以来眼肌、球状肌(面部肌肉、用于说话和吞咽的肌肉)和四肢肌肉的疲劳性无力的病史,类似的亲属患病史,以及各种测试来诊断CMS。
这些检查包括肌电图(EMG)反应减退,以及乙酰胆碱受体(AChR)和肌肉特异性受体酪氨酸激酶(MuSK)的抗体检测阴性。然而,许多CMS患者的家族史是阴性的;在其他患者中,发病时间延迟,EMG异常并非出现在所有的肌肉中,或只是间歇性出现,而且无力的分布是有限的。
肌电图或EMG试验记录了骨骼(自主)肌肉在休息和肌肉收缩时的电活动。递减性肌电图反应是通过以每秒2-3次的速度刺激运动神经来测量的;肌肉的诱发电反应,即复合肌肉动作电位,或CMAPs,由置于受刺激肌肉上方皮肤上的电极记录。如果第四次诱发的CMAP比第一次诱发的CMAP小10%以上,则反应异常。单纤维肌电图是一种更敏感但特异性较低的肌无力障碍测试。在这种测试中,重复刺激单根肌肉内神经纤维,同时记录每次2到4根肌肉纤维的诱发的单纤维动作电位。单个动作电位的时间锁定发射异常增加的变异性是神经肌肉传导缺陷的一个早期指标。
对CMS的具体诊断取决于识别疾病基因和该基因的病理突变。市面上的研究可以很容易地检测出以前确定的CMS类型的突变。通过全外显子组测序或全基因组测序可以检测到以前未被识别的CMS类型的突变,但对获得的结果进行生物信息学分析仍然具有挑战性。CMS的基因诊断非常重要,因为有利于一种类型CMS的治疗可能会使另一种类型的CMS恶化。
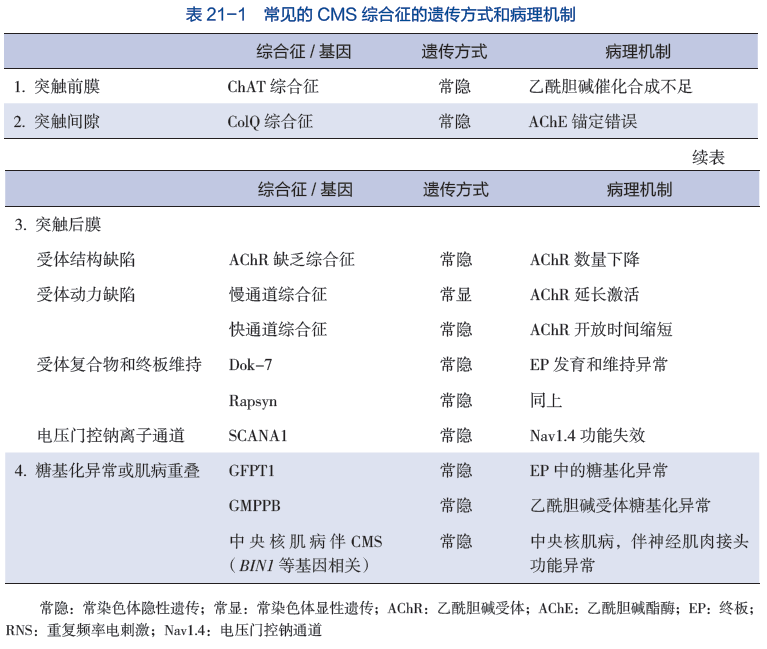
1. 电生理检查 先天性肌无力综合征的重复神经电刺激表现为低频电流重复刺激后出现波幅递减,单纤维肌电图(SFEMG) 可见 Jitter 阻滞和增宽。在 ChAT 突变时,低频重复电刺激(3Hz) 可能缺乏递减反应,此时可予 10Hz 进行延长刺激或在刺激前进行运动诱发,可引出递减反应。ColQ 综合征和慢通道综合征中,因为突触后膜持续兴奋,给予单一电刺激,在第一个 CMAP 波后,出现一个重复 CMAP 波(R-CAMP)(图 21-1)。
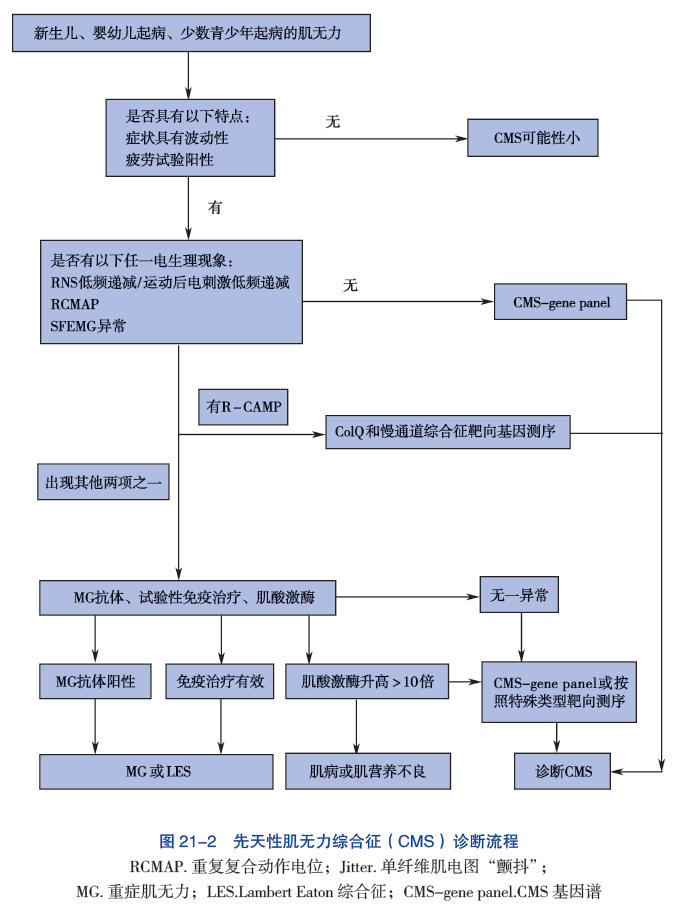
七、治疗
目前还没有针对受影响个体的标准化治疗方案或指南。由于CMS总体上的罕见性,以及某些亚型只在少数人或更少的人身上被发现,因此没有在一大群病人身上进行过治疗试验。医学文献中报道了各种治疗方法,作为单个病例报告或小系列病人的一部分。治疗试验将非常有助于确定特定药物和治疗方法对CMS患者的长期安全性和有效性。
如上所述,确定每个人的具体亚型是非常重要的,因为对一种CMS有效的药物可能对另一种CMS无效,甚至有害。有关CMS具体亚型的更详细的治疗信息,请见上文每个亚型列表下的 "症状和体征 "部分。
目前治疗CMS的药物包括被称为胆碱能激动剂的药物,如吡啶斯的明或阿米夫林(3,4-二氨基吡啶),乙酰胆碱受体离子通道的长效开放通道阻断剂氟西汀和奎尼丁,以及肾上腺素能激动剂如沙丁胺醇和麻黄碱。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
1. Harper CM. Electrodiagnosis of myasthenic disorders. In: Engel AG, ed. Myasthenia Gravis and Myasthenic Disorders. 2nd ed. New York: Oxford; 2012: pp. 37-59.
2. Ohno K, Tsujino A, Shen XM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA. 2001;98:2017-2022.
3.Shen XM, Crawford TO, Brengman J, et al. Functional consequences and structural interpretation of mutations in human choline acetyltransferase. Hum Mutat. 2011;32:1259-1267.
4. Byring RF, Pihko H, Shen XM, et al. Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord. 2002;12:548-553.
5. Maselli RA, Chen D, Mo D, Bowe C, Fenton G, Wollman RL. Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve. 2003;27:180-187.
6. Mallory LA, Shaw JG, Burgess SL, et al. Congenital myasthenic syndrome with episodic apnea. Pediatr Neurol. 2009;41:42-45.
7. Schara U, Christen HJ, Durmus H, et al. Long-term follow-up in patients with congenital myasthenic syndrome due to CHAT mutations. Eur J Paediatr Neurol. 2010;14:326-333.
8. Kraner S, Laufenberg I, Strassburg HM, Sieb JP, Steinlein OK. Congenital maysthenicsyndrome with episodic apnea in patients homozygous for a CHAT missense mutation. Arch Neurol. 2003;60:761-763.
9. Shen XM, Selcen D, Brengman J, Engel AG. Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology. 2014;83:2247-2255.
10. Herrmann DN, Horvath R, Snowden JE, et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of Lambert-Eaton myasthenic syndrome and nonprogressive motor neuropathy. Am J Hum Genet. 2014;95:332-339.
11. Walls TJ, Engel AG, Nagel AS, Harper CM, Trastek VF. Congenital myasthenic syndrome associated with paucity of synaptic vesicles and reduced quantal release. Ann NY Acad Sci. 1993;681:461-468.
12. Ohno K, Engel AG, Brengman JM, et al. The spectrum of mutations causing endplate acetylcholinesterase deficiency. Annals of Neurology. 2000;47:162-170.
13. Bestue-Cardiel M, de-Cabazon-Alvarez AS, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005;65:144-146.
14. Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and DOK-7 myasthenia. Muscle Nerve. 2011;44:789-794.
15. Maselli RA, Ng JJ, Andreson JA, et al. Mutations in LAMB2 causing a severe form of s synaptic congenital myasthenic syndrome. J Med Genet. 2009;46:203-208.
15a. Maselli RA, Arredondo J, Vázquez J, et al. Presynaptic congenital myasthenic syndrome with a homozygous sequence variant in LAMA5 combines myopia, facial tics, and failure of neuromuscular transmission. Am J Med Genet A 2017;173:2240-2245.
16. Engel AG. The therapy of congenital myasthenic syndromes. Neurotherapeutics. 2007;4:252-257.
17. Sadeh M, Shen XM, Engel AG. Beneficial effect of albuterol in congenital myasthenic syndrome with ε subunit mutations. Muscle Nerve. 2011;44:289-291.
18. Engel AG, Ohno K, Sine SM. Sleuthing molecular targets for neurological diseases at the neuromuscular junction. Nature Rev Neurosci. 2003;4:339-352.
19. Harper CM, Fukudome T, Engel AG. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:170-173.
20. Hesselmans LFGM, Jennekens FGI, Vand Den Oord CJM, Veldman H, Vincent A. Development of innervation of skeletal muscle fibers in man: Relation to acetylcholine receptors. Anat Rec. 1993;236:553-562.
21. Morgan NV, Brueton LA, Cox P, et al. Mutations in the embryonal subunit of the acetylcholine receptor ( CHNRG ) cause lethal and Escobar variants of the multiple pterygium syndrome. Am J Hum Genet. 2006;79:390-395.
22. Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol. 2013;5:a009167-a009167.
23. Huze C, Bauche S, Richard P, et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am J Hum Genet. 2009;85:155-167.
24. Maselli RA, Fernandez JM, Arredondo J, et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural agrin (z-) agrin. Hum Genet (Berlin). 2012;131:1123-1135.
25. Nicole S, Chaouch A, Torbergsen T, et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain. 2014;137:2429-2443.
26. Ohkawara B, Cabrera-Serrano M, Nakat T, et al. LRP4 third β-propeller domain mutations cause novel congenital myasthenic syndrome by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. 2014;23:1856-1868.
27. Selcen D, Ohkawara B, Shen, X.-M.,McEvoy, K., Ohno, K. Engel, A.G. Impaired development and maintenance of the neuromuscular junction in LRP4 myasthenia. JAMA Neurology. 2015;72.
28. Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13 3229-3240.
29. Mihaylova V, Salih MA, Mukhtar MM, et al. Refinement of the clinical phenotype in MUSK -related congenital myasthenic syndromes. Neurology. 2009;73:1926-1928.
30. Maselli R, Arredondo J, Cagney O, et al. Mutations in MUSK causing congenital myasthenic syndrome impair MuSK-Dok-7 interaction. Hum Mol Genet. 2010;19:2370-2379.
31. Ben Ammar A, Soltanzadeh P, Bauchˆ S, et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One. 2013;8:e53826.
32. Maggi L, Brugnoni R, Confalioneri P, et al. Marked phenotypic variability in two siblings affected by congenital myasthenic syndrome caused by mutations in MUSK J Neurol. 2013;Epub ahead of print:10/12/2013.
33. Chevessier F, Girard E, Molgo J, et al. A mouse model for congenital myasthenic syndrome due to MuSK mutations reveals defects in structure and function of neuromuscular junctions. Hum Mol Genet. 2008;17:3577-3595.
34. Gallenmuller C, Muller-Felber W, Dusl M, et al. Salbutamol-responsive limb-girdle congenital myasthenic syndrome due to a novel missese mutaion and heteroallelic deletion in MUSK. Neuromuscul Disord. 2014;24:31-35-2014.
35. Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802-1805.
36. Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975-1978.
37. Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71-87.
38. Schara U, Barisic N, Deschauer M, et al. Ephedrine therapy in eight patients with congenital myasthenic syndrome due to DOK7 mutations. Neuromuscul Disord. 2010;19:828-832.
39. Ramarao MK, Cohen JB. Mechanism of nicotinic acetylcholine receptor cluster formation by rapsyn. Proc Natl Acad Sci USA. 1998;95:4007-4012.
40. Ohno K, Engel AG, Shen XM, et al. Rapsyn mutations in humans cause endplate acetylcholine receptor deficiency and myasthenic syndrome. Am J Hum Genet. 2002;70:875-885.
41. Burke G, Cossins J, Maxwell S, et al. Rapsyn mutations in hereditary myasthenia. Distinct early- and late-onset phenotypes. Neurology. 2003;61 826-828.
42. Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: Clinical and molecular findings in 39 patients. Neurology. 2009;73:228-235.
43. Banwell BL, Ohno K, Sieb JP, Engel AG. Novel truncating RAPSN mutation causing congenital myasthenic syndrome responsive to 3,4-diaminopyridine. Neuromuscul Disord. 2004;14:202-207.
44. Skeie GO, Aurlien H, Mller JS, Norgard G, Bindoff LA. Unusual features in a boy with rapsyn N88K mutation. Neurology. 2006;67:2262-2263.
45. Muller JS, Mildner G, Mller-Felber W, et al. Rapsyn N88K is a frequent cause of CMS in European patients. Neurology. 2003;60:1805-1811.
46. Cossins J, Burke G, Maxwell S, et al. Diverse molecular mechanisms involved in AChR deficiency due to rapsyn mutations. Brain. 2006;129:2773-2783.
47. Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-box mutations in RAPSN promoter region in eight cases with congenital myasthenic syndrome. Hum Mol Genet. 2003;12:739-748.
48. Haeuptle MA, Hennet T. Congenital disorders of glycosylation: An update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum Mutat. 2009;30:1628-1641.
49. Freeze HH, Chong JX, Bamshad MJ, Ng BG. Solving glycosylation disorders: Fundamental approaches reveal complicated pathways. Am J Hum Genet. 2014;94:161-165.
50. Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am. J. Hum. Genet. 2011;88:162-172.
51. Selcen D, Shen XM, Milone M, et al. GFPT1-myasthenia: Clinical, structural, and electrophysiologic heterogeneity. Neurology. 2013;23:370-378.
52. Belaya K, Finlayson S, Slater C, et al. Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am J Hum Genet. 2012;91:1-9.
53. Selcen D, Shen XM, Li Y, Stans AA, Wieben E, Engel AG. DPAGT1 myasthenia and myopathy. Genetic, phenotypic, and expression studies. Neurology. 2014;82:1822-1830.
54. Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain. 2013;136:944-956.
55. Regal L, Shen XM, Selcen D, Verhille C, Meulemans S, Creemers JWM. PREPL deficiency with or without cystinuria causes a novel myasthenic syndrome. Neurology. 2014;82:1254-1260.
56. Radhakrishnan K, Baltes J, Creemers JWM, Schu P. Trans-Golgi network morphology and sorting is regulated by prolyl-oligopeptidase-like protein PREPL and AP-1 complex subunit æ1A. J Cell Sci. 2013;126:1155-1163.
57. Kim MH, Hersh LB. The vesicular acetylcholine transporter interacts with clathrin-associated adaptor complexes AP-1 and AP-2. J Biol Chem. 2004;279:12580-12587.
58. Tsujino A, Maertens C, Ohno K, et al. Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. 2003;100:7377-7382.
59. Arnold WD, Feldman DH, Ramirez S, et al. Defective fast inactivation recovery of Nav 1.4 in congenital myasthenic syndrome. Ann Neurol. 2015;77:840-850.
60. Elliott CE, Becker B, Oehler S, Castanon MJ, Hauptmann R, Wiche G. Plectin transcript diversity: identification and tissue distribution of variants with distinct first coding exons and rodless isoforms. Genomics. 1997;42:115-125.
61. Fuchs P, Zorer M, Rezniczek GA, et al. Unusual 5′ transcript complexity of plectin isoforms: novel tissue- specific exons modulate actin binding activity. Hum Mol Genet. 1999;8:2461-2472.
62. Konieczny P, Wiche G. Muscular integrity – a matter of interlinking distinct structures via plectin. In: Laing NG, ed. The sarcomere and skeletal muscle disease: Springer; 2008:165-175.
63. Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76:327-336.
64. Edvardson S, Porcelli V, Jalas C, et al. Agenesis of corpus callosum and optic nerve hypoplasia due to mutation in SLC25A1 encoding the mitochondrial citrate transporter. J Med Genet. 2013;50:240-245.
65. Chaouch A, Porcelli V, Cox DM, et al. Mutations in the mitochondrial citrate carrier SLC25A1 are associated with impaired neuromuscular transmission. J Neuromuscul Dis. 2014;1:75-90.
66. Claeys KG, Maisonobe T, Bohm J, et al. Phenotype of a patient with recessive centronuclear myopathy and a novel BIN1 mutation. Neurology. 2010;74:519-521.
67. Robb SA, Sewry CA, Dowling JJ, et al. Impaired neuromuscular transmission and response to aceylcholinesterase inhibitors in centronuclear myopathy. Neuromuscul Disord. 2011;21:379-386.
68. Gibbs EM, Clarke NF, Rose K, et al. Neuromuscular junction abnrormalities in DNM2-related centronuclear myopathy. J Mol Med (Berl). 2013;91:727-737.
69. Liewluck T, Shen XM, Milone M, Engel AG. Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord. 2011;21:387-395.
70. Engel AG, Shen,X.M., Sine,S.M. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurology. 2015;14:420-434.
71. Engel AG. Anatomy and molecular architecture of the neuromuscular junction. In: Engel AG, ed. Myasthenia Gravis and Myasthenic Disorders. 2nd Ed. New York: Oxford University Press; 2012 pp 1-36.
72. Engel AG. Congenital myasthenic syndromes. In: Engel AG, ed. Myasthenia Gravis and Myasthenic Syndromes. 2nd Ed. New York: Oxford University Press; 2012, pp 173-230.
73. https://www.chard.org.cn/#/knowledge/jbzsk/detail/41
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#诊断与治疗#
84
#先天性#
70
#流行病#
72
#综合征#
79
#肌无力#
56