
RNA-seq方法原理、数据分析、数据库及工具全面介绍
2014-01-04 MedSci MedSci原创

bsp;- De novo assembly of short reads with robust error correction. An improvement on early versions of SSAKE. * Velvet - Velvet is a de novo genomic assembler specially designed for sho
能够对RNA序列数据进行分析的新方法可以让我们从头开始构建转录组。
对
RNA进行测序一直以来都被认为是一种发现基因的有效方法,而且这种方法还被认为是对编码基因以及非编码基因进行注释的金标准。与以前的方法相比,大规模
平行RNA测序方法(massively parallel sequencing of
RNA)极大增强了RNA测序技术的处理能力,使我们得以能够对转录组进行测序。在本文中即将介绍到的这两种RNA测序方法就能以前所未有的精度对转录组
进行分析。Trapnell小组使用的方法是一种名为Cufflinks的软件。这种软件能够随时发现小鼠生肌细胞(myoblast
cell)内新出现的转录子,还能在细胞分化时对转录子表达水平进行监测,从而分析基因表达情况和剪接情况。Guttman小组也使用了与
Trapnell小组相类似的软件方法,不过他们使用的是另一种名为Scripture的软件。Scripture软件可以对源自三个小鼠细胞系的转录组
进行再注释(reannotate),从而对数百个最近新发现的lincRNA(large intergenic noncoding
RNA)进行完整的基因模式注释。
虽然RNA测序技术已经出现了将近20年,但直到最近才开始构建克隆文库。对人类、小鼠以及其它重要模
式生物进行全长基因克隆构建的科研项目需要几年的时间才能够完成。但是有了最新的测序技术,我们将不再需要构建克隆文库,可以直接对cDNA片段进行测
序。我们现在可以只需要花费几天,仅用以往同类项目科研经费的很少一部分就能够得到一个比较满意的完整的细胞转录组。但是这种新技术也存在一点问题。不用
构建克隆,我们就无法知道哪一个“结果(mRNA或蛋白)”来自哪一个转录子。最近已经有人开始通过对已知的或者预测出来的转录子的短RNA序列进行测序
的方式来对基因表达和可变剪接进行分析研究。虽然这些研究可以得到很多信息,但是这种方法只能用于分析已知基因和对已知的可变连接区域进行分析。为了充分
利用RNA序列数据进行生物学研究,我们还应该能够重建转录子并且还要能够在不借助参考注释基因组信息的情况下对这些转录子的相对丰度进行精确的测量。
过
去我们在利用短RNA序列重建转录子时主要采用了两条策略(图1)。第一条策略是利用ABySS软件从头构建的方法,这样就可以与全长cDNA序列进行比
对,从而解决序列注释的问题。这种办法还可以用于发现参考基因组中未收录或者收录不完全的转录子,还可以用于发现那些缺乏参考基因组RNA序列数据物种的
转录子。不过这种利用小片段序列从头组装转录子的方法实施起来非常困难,只有丰度很高的转录子才有可能被成功组装。
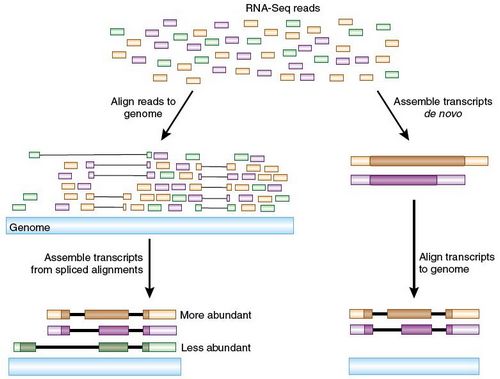
RNA-Seq reads:短片段RNA序列;Align reads to genome:与基因组数据比对;Genome:基因组;Assemble transcripts de novo:从头组装转录子;More abundant:高丰度;
Assemble transcripts from spliced alignments:通过与各种剪接方案比对组装转录子;
Align transcripts to genome:将转录子与基因组进行比对;Less abundant:低丰度;
图1 利用RNA序列数据重建转录子的两种方法。
图
中左侧示意的先比对再组装的办法是Trapnell小组和Guttman小组采用的方法。该方法首先将短片断RNA序列与基因组序列进行比对,计算出所有
可能的剪接方案,然后根据这些剪接方案重建出转录子。图中右侧展示的则是先组装再比对的方法。该方法先从根据RNA片段序列直接头合成出转录子序列,然后
再用各种剪接方式对合成的转录子进行剪接,将剪接产物与基因组进行比对,找出内含子和外显子结构,以及各个不同剪接体之间的差异。由于这种从头合成的方法
绝大部分情况下只对高丰度转录子管用,因此左侧图中展示的先比对再组装的策略要更为灵敏,不过这一观点尚需进一步论证。图中每个RNA片段都根据其来源转
录子被标上了各种颜色。重建转录子中的蛋白编码区被标记成了深色。
第二种策略是先将每一个短片段RNA与基因组进行比对,然后再重建转录
子。Trapnell小组和Guttman小组采用的就是这种策略。这两个小组使用的都是TopHat比对软件,通过该软件与基因组进行比对,获得了大量
的剪接体。早期的RNA测序只能得到25~32个碱基长度的序列片段,现在我们可以得到75个碱基甚至更长的序列片段,这样就更容易进行序列比对,可以将
片段末端固定在不同的外显子当中来判断哪种剪接体才是正确的,这样就不需要借助先前的注释信息了。通过上述这两种方法最终都能得到各种转录子图谱,再通过
末端配对信息剔除掉不太可能的选择最终就能得到想要的转录子。
在使用哪种算法方面也是有很大差别的。比如Trapnell小组采用的
Cufflinks软件就使用了一种非常严格的算术模型来发现每一个位点所有的可变调控转录子,还可以计算出每一种转录子的优势度。Guttman小组采
用的Scripture软件则采用了统计学分段模型(statistical segmentation
model)来区分表达位点和实验噪声。需要对Cufflinks软件技术、Scripture软件技术以及利用ABySS软件从头构建的方法进行大规模的测试,才能判断出哪一种方法在哪一种情况下面最为合适、有效。
令
人吃惊的是,尽管我们已经利用数以百万计的EST和数千条完整的全长cDNA序列对小鼠基因组进行了详细的注释工作,但是Trapnell小组和
Guttman小组还是发现了数千条以前从未发现过的转录子,其中包括已知基因的新型同工型转录子以及全新的编码基因及非编码基因的转录子。
Trapnell
小组发现了3724条新的可信度非常高的已知基因的同工型转录子,这些转录子不论在人工注释的基因数据库还是在自动注释的基因数据库中都没有收录过。
Trapnell小组还发现他们所发现的每一个转录子经过单独的表达验证之后都能对后续的分析起到重要的作用。实验表明,RNA测序工作能够在很大一个动
态范围内准确地反映基因的表达情况,但是之前的实验都只能根据已知的同工型转录子或者预测的同工型转录子来进行判断。根据RNA片段的测序结果直接重建出
所有的同工型转录子,然后再根据这些同工型转录子的出处将所有的配对片段进行分类,Trapnell小组用这种方法就能非常准确地判断出每一个基因的每一
个同工型转录子的表达水平。他们还发现将每一个RNA片段正确地组装入转录子,能够极大的影响同一基因其它已知同工型转录子的预计表达水平。
如
果能够检测每一个同工型转录子的表达水平,那么我们就能够对基因表达的调控机制进行更加深入的研究。这种调控机制可能发生在转录水平,比如形成具有不同转
录起始位点的同工型转录子;也可以发生在转录后水平,比如虽然转录起始位点相同,但是内部剪接方式不同的各同工型转录子。Trapnell小组还发现,随
着实验的推进,有大量基因的表达都会因为上面提到的这两种调控机制而发生明显的改变。这种能够在如此长时间段里对整个基因组表达调控情况进行检测的能力让
我们能够进一步了解到基因组的新功能。比如,在这种水平上的详细数据能够让我们构建出更加合适的基因组调控网络模型,也可以利用这些数据根据每个基因各同
工型转录子剪接情况与表达情况之间的关系来改变模型中的某些调控参数,而不用直接改变每一个基因的参数。
Guttman小组也发现了很多
新的同工型剪接转录子,不过他们的工作重点主要都放到了各个新发现的转录子身上,尤其是lincRNA。之前利用芯片测序(ChIP-Seq)方法和全基
因组瓦片芯片(whole-genome tiling
array)方法发现了编码lincRNA的位点,但是由于分辨率不够因此不能构建出准确的模型。Guttman小组在Scripture软件的帮助下对
609个已知位点构建出了基因模型,同时还发现了1000多个新的lincRNA,并解析了这些lincRNA的结构。Guttman小组还发现了469
个蛋白编码基因的反义转录子。
通过为这些非编码RNA构建基因模型的方式能够让我们更好地开展基因功能研究了。比如Guttman小组就
检测了各转录子的保守情况。与以前的观察结果一样,lincRNA要比内含子序列保守得多,但是其保守程度不如蛋白编码序列高。相反,反义转录子并不比编
码蛋白的外显子区域的保守水平高,这说明这两种转录子各自具有不同的功能。RNA测序数据还能够展示非编码转录子的表达模式,这些数据表明lincRNA
的丰度不仅要比蛋白编码RNA的丰度低,同时其表达水平也较低,而且同蛋白编码RNA相比,lincRNA的表达还具有非常明显的组织特异性。简单来说,
如果能够更详细地了解非编码RNA的表达模式,为这些RNA构建出更为准确的基因模型,那么我们就能够更加清楚地知道它们在基因表达调控网络模型以及基因
间相互作用模型中的作用,从而对它们的功能有更加深入的了解和认识。
Trapnell小组和Guttman小组发现了如此之多的新转录子
这一事实不由得不让我们思考一个问题,为什么我们的注释工作会如此滞后呢?在Trapnell小组的试验中,已知的各种同工型转录子占到了总数的80%以
上,这说明这些已知的转录子都来自高表达水平的基因,因此很容易通过以往的cDNA克隆测序方法给发现。Guttman小组的情况也基本相同。还有11%
的RNA片段是来自已知基因新发现的同工型转录子,其中62%的片段都能与现有的EST或mRNA相印证,但是它们都还没有作为一个独立的转录子被注释。
可能在以往的研究当中也零星的发现过这些低丰度的同工型转录子,可能只是因为它们与已知的转录子比较相似,或者是因为没能得到完整的测序,因此没有进行注
释。与这种情况类似,被Guttman小组发现的lincRNA中有43%都可以在以往的小鼠cDNA研究工作中找到踪迹。由于lincRNA具有明显的
组织特异性而以往的研究工作往往又只局限于研究某些组织,因此剩余的57%的lincRNA应该都是以前没有发现过的新的转录子。早期大规模RNA测序工
作的重点都是针对蛋白编码区域,这可能也是我们注释工作显得落后的原因之一。Trapnell小组和Guttman小组采用的这种RNA测序方法能够明白
无误地区分编码转录子和非编码转录子。
Trapnell小组使用的Cufflinks软件、Guttman小组使用的Scripture
软件,以及其它一些类似的软件可以极大地改进我们的基因组注释工作,不论是被研究得非常详细的基因组还是缺乏EST和全长mRNA序列信息的基因组都能从
中受益。但是利用RNA测序方法来进行基因注释工作也不是完美无缺的。通过Cufflinks软件和Scripture软件被发现的转录子中有大量的转录
子都属于已知的转录子,只不过因为覆盖率较低所以都是不完整的转录子。正如用RNA测序方法重建的转录子很难与EST数据相吻合一样,很多低表达水平或者
组织特异性表达的转录子也很难通过现有的RNA测序方法被发现。
随着测序技术的不断进步,我们也能够对转录组开展更为深入的测序工作,能
够发现更多、更可靠的转录子。不过我们还需要更加先进的方法来区分低丰度的功能性转录子和背景噪声以及各种人为的假象。虽然Cufflinks和
Scripture都是非常好的基因组注释工具,但针对不同的基因组(因为每个基因组的特点比如基因的密度、内含子的长度和含量、可变剪接发生的频率高低
等等都不尽相同),我们仍然需要各种不同的软件(或算法)来更好地匹配并注释这些基因组。我们还需要看看Cufflinks和
Scripture在处理其它与小鼠基因组完全不同的基因组时表现如何。
大规模并行测序技术已经彻底改变了我们对基因组的研究方法,测序
结果的质量也在不断提高,得到的信息量也在爆炸式增长。通过本文的介绍,我们也可以看到RNA测序技术以及转录子发现技术对于基因组注释工作以及基因组转
录水平及转录后水平调控机制研究工作的重要意义。如果相应的软件能够及时跟上,那么RNA测序技术将有更大的成就。
原文检索:
Brian J Haas & Michael C Zody. (2010) Advancing RNA-Seq analysis, Nature Biotechnology, 28(5): 421-423.
{nextpage}
单细胞RNA-seq方法的比较
随着大家对单细胞转录组分析的兴趣日益增加,开展单细胞RNA-seq的方法也在不断涌现。近日,斯坦福大学的研究人员比较了市场上各种单细胞RNA扩增方法,以系统评估单细胞RNA-seq方法的灵敏度和准确性。他们的研究结果发表在Nature Methods在线版上。
目前,整个转录组的高通量测序(RNA-seq)已广泛用于各种组织的基因表达分析。然而,人们开展单细胞转录组分析的愿望也越来越强烈,这是因为有些细胞量少珍贵,不足以开展传统的RNA-seq,也有人希望对异质群体中的细胞亚群开展分析。过去的基因表达“平均值”已不再能满足人们的需求。
近年来,开展单细胞RNA-seq的各种方法时有报道,但关于这些方法的通量和性能还存在一些问题。比如说,性能主要是通过灵敏度和精确性来评估的。然而,为了评估测定方法的有效性,还应当评估准确性,即测量值与真实值之间有多接近。
为 此,斯坦福大学生物工程系的Stephen R Quake领导的研究小组对102个人类单细胞转录组进行了定量的RNA-seq分析。他们采用了三种单细胞RNA扩增方法,分别为SMARTer Ultra Low RNA Kit(Clontech)、TransPlex Kit(Sigma-Aldrich)以及利用C1微流体系统(Fluidigm)进行SMARTer cDNA合成。同时,他们也利用大量总RNA开展传统的RNA-seq,并通过多重qPCR对同一细胞类型中的40个基因进行独立定量,来评估单细胞方法的准确性。
由 于很难获得绝对表达水平,研究人员计算了每种细胞的相对表达水平,即相对所有转录本中位数表达的基因表达。所产生的无量纲数应独立于测定方法,可直接比 较,以确定方法的准确性。他们发现,对于单细胞方法,qPCR和RNA-seq的表达值相关性良好(r > 0.84),证实了单细胞RNA-seq方法能够以定量的方式检测基因表达,与目前的金标准qPCR方法一致。
有趣的是,各种方法之间的相关系数和线性回归的斜率不同。相关系数表示每种方法能够在多大程度上再现qPCR所测定的表达,而斜率表示一些失真。他们发现, 以纳升体积开展样品制备(C1系统)产生了几乎为1的回归斜率,表明绝对准确性最高。一种可能的解释是,减小反应室的体积可增大反应物的有效浓度,减少模 板与非特异分子之间的竞争,从而减少扩增偏向。
此外,研究人员还直接比较了微升和纳升级的样品制备,发现对于qPCR和RNA-seq,以纳升体积制备的样品都产生了更少的假阳性。作者认为,与管式的样品制备相比,C1平台表现出一些优势,包括假阳性低和偏向小。
作 者总结道,他们的结果表明,单细胞RNA-seq可产生与qPCR相当的结果,特别是在以纳升反应体积制备样品时(如微流体装置C1)。通常在样品制备过 程中引入的偏向减小,而相关性也进一步提高。他们希望,低偏向、高通量的单细胞RNA-seq将让原代组织的研究变得常规。{nextpage}
RNA-seq数据差异表达分析方法的比较
BMC Bioinformatics 2013, 14:91 doi:10.1186/1471-2105-14-91
A comparison of methods for differential expression analysis of
RNA-seq data
Charlotte Soneson (Charlotte.Soneson@isb-sib.ch)
Mauro Delorenzi (Mauro.Delorenzi@unil.ch)
“发现条件间差异表达的基因是理解表型变异的分子基础的一个有机部分。过去几十年中,DNA微阵列被广泛用于定量不同基因的mRNA丰度,更近期的 RNA-seq作为一个强有力的竞争者冒了出来。随着测序成本持续下降,可以想象使用RNA-seq做差异表达分析会迅速增加。为了探索可能性和解决这种 相对新型的数据提出的挑战,大量软件包特别为RNA-seq数据的差异表达分析开发出来了。”
而本文的结果是:
“我们广泛比较了RNA-seq数据的差异表达分析的7种方法。所有方法都可以在R框架下免费获得,并以一个计数矩阵作为输入,计数即每个样品中映射到每个感兴趣的基因组特征上的reads数目。我们基于模拟数据和实际RNA-seq数据评价了这些方法。”
结论就是:
“极小样本量仍是RNA-seq实验的普遍状况,对所有评价方法造成了困难;而任何在这样的条件下获得的结果都应该谨慎解释。对于更大的样本量,组合稳定方差变换和limma方法来进行差异表达分析会在很多不同的条件下表现良好,正如非参数的SAMseq方法一样。”
到2013年还说这种话,这些结论实在有点鸡肋啊~ 貌似为SAMseq摇旗呐喊来的……不过:
比较了11种软件包,这还是前所未有的:DESeq、edgeR、NBPSeq、TSPM、baySeq、EBSeq、NOISeq、SAMseq、
ShrinkSeq这9种可直接处理计数数据,另两种分别是voom(+limma)和vst(+limma),转换数据后用limma做差异表达分析。
正如很多文章已经提到的那些,RNA-seq比起微阵列有三大优点:
1、更大的动态范围
2、更低的背景噪音
3、能检测和定量先前未知的转录本及亚型
RNA-seq也有一些难题:
1、样本内不均匀性:基因组区域之间核苷酸组成的变异性导致沿基因组的read覆盖深度并不均匀;
2、同样表达水平下,长基因比短基因有更多的reads;
3、对于条件之间的表达差异,分别对各个基因进行差异表达分析,而忽略了样本内的偏倚(它们被假设对所有样本有类似的影响)
4、样本间不均匀性:测序深度或文库大小
5、少数高表达基因抑制了其他基因的read计数比例,可能导致低表达基因的差异表达假阳性
1、对上述4,5两点,估计样本特异的归一化因子,用于重新调整观测计数。这些归一化方法是为了使样本间的非差异表达基因的归一化计数是相似的。本研究中 使用的是DESeq包中的TMM方法。归一化因子和TMM两种方法的性能相似,也是仅有的两个在文献9的评价中对所有度量都提供了满意结果的方法。
2、对于2、4两点,基因长度和文库大小,有的软件采用了RPKM或相关的FPKM方法。只有非参数方法才适用于RPKM值。
致命的假设:大部分基因在样本间的表达是相等的。于是差异表达基因分成上调、下调及其之间的或多或少相等的三部分。
差异表达已经提出的模型中,Poisson分布和负二项分布最常见,还有β分布也被提出来。Poisson分布很适合技术变异的分析;而更高的生物学变异需要合并过离散的分布,如负二项分布。
RNA-seq数据的差异表达分析仍处于它的婴儿期,需要不断提出新的方法来。目前没有一致认可的表现最好的方法,只发表了很少的现有方法的广泛比较。文
献19中,依照区分真实差异/非差异表达基因的能力比较了四种参数方法。还有研究评价了测序深度的影响,并与样本量进行了比较,并发现后者的影响相当大。
本文中比较了为不同条件下RNA-seq数据的差异表达分析开发的11种方法。其中9种直接对计数数据进行建模,而另两个先对计数进行变换再应用微阵列数
据的差异表达分析的传统方法。研究限于R框架下实现的可应用于计数矩阵的可用方法。进一步我们聚焦于发现两条件之间的差异表达基因,因为这是最常见的应
用,虽然大多数方法也允许更复杂的试验设计。
对NB和Poisson分布模拟的数据和分别加了一些例外点的数据共四种数据集,研究了在不同实验条件下方法的下列方面:
1、排序真实DEGs在nonDEGs之前的能力;
2、在给定水平控制I型错误和假发现率的能力;
3、计算时间。
对于真实RNA-seq数据,比较了DEGs集,各自的数目和重叠的数目。还研究了不同方法获得的基因排序的一致性。
六种方法有名义p-value(edgeR、DESeq、NBPSeq、TSPM、voom+limma、vst+limma),我们定义分数值为1-
pvalue。对于SAMseq,定义平均Wilcoxon统计量的绝对值为排序分值,而对于baySeq、EBSeq、ShrinkSeq使用估计的差
异表达后验概率,或者等价地,1-BFDR,其中BFDR表示估计的Bayesian
FDR。对于NOISeq,使用统计量q_NOISeq。所有这些分数都是双侧的,即不被差异表达的方向影响。给定上述分数的阈值,我们就选择出阈值以上
的为DEGs,其余的为non-DEGs。
SAMseq使用了重抽样策略使文库大小一致,因此隐含假设了所有归一化因子是相等的,在整体上调的模拟研究中表现最好。上下调基因都有的时候,所有方法 的AUC(Area under the ROC curve (AUC),Receiver Operating Characteristic)性能类似。TSPM和EBSeq在所有方法中表现出最强烈的样本量依赖性,其次是SAMseq和baySeq。对于最小样 本量(每条件下2样本),最佳结果是DESeq、edgeR、NBPSeq、voom+limma和vst+limma。
当所有DEGs上调时,baySeq结果的变异性很高;而DEGs向不同的方向调整时这种变异就会减小。
将真实DEGs排的很靠前的方法是基于变换的voom+limma和vst+limma方法和ShrinkSeq,但是TSPM和NOISeq还会把一些
真实的nonDEGs排的很靠前。SAMseq也表现不错,但是有一些真实DEGs和nonDEGs被返回靠前的相同的值。
……实在是相当繁琐,直接看结论吧……
Conclusions
文本评价和比较了11种RNA-seq数据的差异表达分析方法。主要结果如下:
Table 2 Summary of the main observations
DESeq - Conservative with default settings.
Becomes more conservative when outliers are introduced.
- Generally low TPR.
- Poor FDR control with 2 samples/condition, good FDR control for
larger sample sizes, also with
outliers.
- Medium computational time requirement, increases slightly with
sample size.
edgeR - Slightly liberal for small sample sizes
with default settings. Becomes more liberal when outliers are
introduced.
- Generally high TPR.
- Poor FDR control in many cases, worse with outliers.
- Medium computational time requirement, largely independent of
sample size.
NBPSeq - Liberal for all sample sizes. Becomes
more liberal when outliers are introduced.
- Medium TPR.
- Poor FDR control, worse with outliers. Often truly non-DE genes
are among those with smallest p-
values.
- Medium computational time requirement, increases slightly with
sample size.
TSPM - Overall highly sample-size dependent
performance.
- Liberal for small sample sizes, largely unaffected by
outliers.
- Very poor FDR control for small sample sizes, improves rapidly
with increasing sample size.
Largely unaffected by outliers.
- When all genes are overdispersed, many truly non-DE genes are
among the ones with smallest p-
values. Remedied when the counts for some genes are Poisson
distributed.
- Medium computational time requirement, largely independent of
sample size.
voom / vst
- Good type I error control, becomes more conservative when
outliers are introduced.
- Low power for small sample sizes. Medium TPR for larger sample
sizes.
- Good FDR control except for simulation study B04000. Largely
unaffected by introduction of outliers.
- Computationally fast.
baySeq - Highly variable results when all DE genes
are regulated in the same direction. Less variability when the DE
genes are regulated in different directions.
- Low TPR. Largely unaffected by outliers.
- Poor FDR control with 2 samples/condition, good for larger sample
sizes in the absence of outliers. Poor FDR control in the presence
of outliers.
- Computationally slow, but allows parallelization.
EBSeq - TPR relatively independent of sample size
and presence of outliers.
- Poor FDR control in most situations, relatively unaffected by
outliers.
- Medium computational time requirement, increases slightly with
sample size.
NOISeq - Not clear how to set the threshold for
qNOISeq to correspond to a given FDR threshold.
- Performs well, in terms of false discovery curves, when the
dispersion is different between the
conditions (see supplementary material).
- Computational time requirement highly dependent on sample
size.
SAMseq - Low power for small sample sizes. High
TPR for large enough sample sizes.
- Performs well also for simulation study B04000.
- Largely unaffected by introduction of outliers.
- Computational time requirement highly dependent on sample
size.
ShrinkSeq - Often poor FDR control, but allows
the user to use also a fold change threshold in the inference
procedure.
- High TPR.
- Computationally slow, but allows parallelization.
没有哪种单独的方法对所有情形都是最优的,特定情形下方法的选择取决于实验条件。本文评价的这些方法中,基于稳定方差的变换与limma组合的方法在很多 情况下都表现不错,而且不受例外点影响、计算很快,但是要求每条件下至少3个样本来提供充分的检定力。而且在两条件下散度不同时表现更糟糕。非参数方法 SAMseq在大样本量时是性能最优的方法,需要至少每条件下4-5个样本提供充分的检定力。对于高表达基因,SAMseq的统计显著性所需的倍数变化比 很多其他方法要低,这可能潜在地折中了一些统计显著的DEGs的生物学显著性。对ShrinkSeq也是一样,不过它有一个选项在推断过程中强加一个倍数 变化要求。
小样本导致一些方法的误报率远超FDR阈值。对于参数方法,这可能是因为均值和方差估计不精确。TSPM受样本量影响最大,可能因为它使用了渐进估计。尽 管发展指向大样本量,而且barcoding和multiplexing创造了固定成本分析更多样本的机会,但是目前为止RNA-seq实验仍然太贵而不 允许广泛的重复。本研究所传达的结果强烈建议小样本差异表达基因应该谨慎解释,真实FDR可能超出所选FDR阈值数倍。
DESeq、edgeR和NBPSeq基于类似的原理,因此基因排序的精确度很类似。但是相同阈值选取出的DEGs有很大不同,这是因为它们估计散度参数 的方法不同。在缺省设置和合理的大样本量下,DESeq通常过于保守而edgeR和NBPSeq通常过于慷慨而得出大量假DEGs。分析表明参数选择影响 很大,而且缺省推荐参数事实上选择的很好通常能得到最佳结果。
EBSeq、baySeq、ShrinkSeq使用了不同的推断方法来估计每个基因差异表达的后验概率。baySeq一些条件下表现不错,但是高度可变, 特别是所有基因都上调或都下调时。大样本量条件下有异常值时,EBSeq比baySeq的假阳性低,小样本量时baySeq比EBSeq的假阳性低。
{nextpage}
从RNA-seq reads到差异表达结果
Alicia Oshlack, Mark D Robinson, Matthew D Young. From RNA-seq reads to differential expression results. Genome Biology 2010, 11:220 http://genomebiology.com/2010/11/12/220
理解RNA-seq数据依赖于感兴趣的科学问题。例如,决定等位点表达的差异需要精确确定转录的SNPs的广泛存在。另一方面融合基因或癌症样品中的畸变 可以通过寻找RNA-seq数据中的新转录本来检测。过去一年(即2009年),一些方法涌现出来,用RNA-seq数据进行丰度估计,可变剪接、RNA 编辑和新转录本的检测。然而,很多生物学研究的基本对象是样品间的基因表达谱。因此,在本评论中我们聚焦于可以检测样品间基因表达水平差异的可用方法。这 种分析与控制实验尤其相关,如比较同一组织的野生型和突变株的表达,比较处理与未处理的细胞,癌症的和正常的细胞等等。我们在此列出用于检测RNA- seq差异表达的处理流程,并检查可以执行该分析的可用方法和开源软件。我们还突出了需要进一步研究的一些方面。
多数RNA-seq实验取一个纯化了的RNA样品,切碎,转换成cDNA并在高通量平台如Illumina GA/HiSeq、SOLiD或Roche 454上测序。该过程产生了来自cDNA片段的一端的、数以百万计的reads(25~300bp)。该过程一个常用的变式是生成双末端reads,即paired-end reads。各平台在化学和处理步骤上有本质不同,但忽略精确的细节后,原始数据都是由带有质量值的短序列的一个长列表组成;这就形成了本评论的切入点。
图1列出了差异表达分析的典型RNA-seq流程概览。首先,reads映射到基因组或转录组。其次,每个样品映射的reads依实验目的而组装成基因水
平、外显子水平或转录本水平的表达概括。接下来,汇总的数据进行归一化以与差异表达的统计检验相协调,产生了一个带有P-value和倍数变化的、排好序
的基因列表。最后,执行系统生物学方法从这些列表中获得生物学的见解,就像在微阵列上进行的那样。我们批判了下列目前可用的RNA-seq数据分析方法的
每一步。我们聚焦于普遍可用的开源软件而不是提供一个所有工具的完整列表。
映射
为了用RNA-seq数据比较样品间表达水平,必须把短reads转换成表达定量。这个过程的第一步就是read映射或比对。最简单的,映射工作就是找到 短read与参考序列已知的唯一位置。然而,真实情形中参考序列从来不是所测序RNA的实际生物源的完美表示。除了样品特异的属性如SNPs和 indels之外,还要考虑来自剪接过的转录组而非基因组的reads。而且,短read有时完美地比对到多个位置,也可能包含不得不考虑的测序错误。因 此,真正的任务是找到短read最佳匹配到参考序列的位置,其中允许错误和结构变异。
尽管对如何最佳比对reads到参考序列的研究还在进行,但是所有的解决办法都涉及在算法的计算需求和允许匹配参考序列的模糊性之间一定的妥协。几乎所有 的短read比对器都采用了首先通过启发式匹配的策略,这迅速找打了可能位置的一个简化列表,接着对候选位置进行全面评估,通过一个复杂的局部比对算法。 如果不做预先的启发式搜索来约减潜在的比对位置数,那么在目前的硬件上执行百万级短reads的局部比对会是计算上不可能的。
目前的比对器能用hash表或Burrows Wheeler变换(BWT)进行快速启发式匹配。hash表比对器对于检测read和参考序列的复杂差异有易于扩展的优点,在不断增加计算需求的代价 下。而BWT比对器可以很有效地映射很接近匹配参考序列的reads,但是一旦考虑更复杂的错配就会大幅度慢下来。这些技术的详细说明可参考文献 23,26-30.
比对器在怎样处理多映射方面也很不一样。多数比对器要么忽略多映射、随机定位它们,要么基于局部覆盖度的估计来定位,尽管结合比对分数的方法也已经提出。PE reads减少了多映射问题,使得多映射的模糊性在大多数情况下都可以解决。
当考虑reads来自基因组DNA时,所有要做的就是映射到一个相关的参考基因组上。但是,RNA-seq是测序转录组片段。这个差异可以用几种方式处 理。既然转录组是建立在基因组之上,那么最常用的(至少是最初的)方法就是用基因组自身做参考序列。这有容易而不偏向任何已知注释的好处。但是跨外显子边 界的reads不会映射到参考序列上。因此,用基因组做参考序列会给出较少外显子的转录本以更高的覆盖度。越长的reads越可能跨外显子边界,因此引起 接合reads比例增加。
为考虑接合reads,通常的实践是建立外显子接合位点库,其中用注视了的外显子的边界构建了参考序列。为了不依赖现存注释的跨外显子边界,可用数据集自 身来从头检测剪接位点。另一个选择是转录组从头组装。所有的从头方法都能鉴定新转录本,并且对没有参考基因组或注释的物种来说是唯一的选项。但是从头方法 是计算密集的,需要长PE reads和高覆盖度以可靠地进行计算。
常用的转录组映射方法是逐渐增加映射策略的复杂性以处理未比对上的reads。
汇总映射的reads
已经尽可能多的获得了reads的基因组位置,下一步任务就是在一些生物学意义单位上汇总和合计这些reads,如外显子、转录本、基因等水平。最简单的 最常用的方法是计数与基因的外显子重叠的reads。但是有相当部分reads映射到基因组上已注释外显子以外的区域,即使是良好注释的物种,如小鼠、 人。
一个可选的汇总是包含沿基因全长的reads,从而结合了内含子reads。这就在汇总中包含了未注释的外显子并考虑了注释不太好或可变的外显子边界。但
是,包含内含子也会捕获到重叠转录本——它们共享一个基因组位置但是源于不同基因。还有其他很多可能的变体用于汇总,如只包含映射到编码序列的reads
或者只汇总从头预测的外显子的reads。接合reads也可添加到基因汇总计数中或用于对剪接亚型的丰度进行建模。这些不同的可能性在图2b中图示说
明。在这些选项下,汇总的选择可能大幅改变每个基因的reads计数,甚至比映射策略的选择影响要大。尽管如此,很少有研究实现了哪种汇总方法是最适合差
异表达检测的。
归一化
归一化使得可以比较样品间和样品内的表达水平。已经证明,归一化是RNA-seq数据的差异表达分析的一个关键步骤。文库内和文库间比较的归一化方法是不同的。
文库内归一化允许定量每个基因相对于样品内其他基因的表达水平。因为越长的转录本有越高的reads计数,文库内归一化的常用方法是用基因长度去除汇总计 数。广泛使用的RPKM在样品内比较中同时解释了文库大小和基因长度的影响。为了验证该方法,Mortazavi等引入了一些阿拉伯芥RNAs到小鼠的组 织样品中,跨过一系列基因长度和表达水平。这些非天然的RNAs称为spike-ins,说明了RPKM给出了基因间表达水平的精确比较。然而,已经证明 表达的转录本的read覆盖深度是不一致的,因为序列内容和RNA制备方法,如随机六聚体引发。把这些认识结合到文库内归一化方法中可能会改进比较表达水 平的能力。使用RNA-seq数据来估计样品中转录本的绝对数目也是可能的,但是这需要RNA标准品和额外信息,如总细胞数和RNA制备产出率。
在样本间检验单个基因的差异表达时,技术偏倚如基因长度与核酸组成,大部分会抵消,因为用于汇总的基础序列在样本之间是相同的。然而,样本间归一化对于相
对不同的文库的比较计数来说仍是必要的。最简单最常用的归一化通过文库的总reads进行调整,考虑了测序深度的影响。但是已经证明需要更聪明的归一化来
考虑组成的影响,或这说事实上小部分高表达基因会占总序列数的相当部分。为了对这些特征进行说明,可以从数据中估计出尺度因子,用于差异表达检验的统计模
型。对于后续分析来说,尺度因子比原始计数有优势。另一方面,分位数归一化和一种用匹配指数律分布的方法也被提出用于RNA-seq的样本间归一化。这些
变换的非线性去除了数据的计数本质,使得不清楚怎样合适地进行差异表达检验。目前,分位数归一化似乎并未改善差异表达检测到合适的尺度因子那样的程度,也
不清楚指数律分布应用于所有数据集的情况。
差异表达
差异表达分析的目标是突出在不同实验条件下丰度显著变化的基因。一般地,这意味着得到每个文库的汇总计数数据表并进行感兴趣样本间的统计检验。
很多方法开发出来以进行微阵列数据的差异表达分析。然而,RNA-seq给出每个基因的离散度量而微阵列的强度给出了一个连续的强度分布。尽管微阵列的强 度通常是对数变换过的,而作为正态分布的随机变量进行分析,但是计数数据的转换并不能用连续分布很好的逼近,特别是对低计数范围和小样本。因此,合适于计 数数据的统计模型对于抽取RNA-seq数据的大部分信息是很重要的。
一般地,Poisson分布构成了对RNA-seq数据进行建模的基础。在任何使用单RNA源的RNA-seq研究中,在一个Illumina GA测序仪的多个lane上进行测序,拟合优度检验表明多数基因在lane之间的分布事实上是Poisson分布的。这一点被独立的技术试验验证过,而且 已有可用的软件工具执行该分析。但是Poisson假设并未很好滴捕捉到生物学变异。因此对于有生物学重复的数据集进行基于Poisson的分析将倾向于 高的假阳性率,源于低估了取样误差。尽管RNA-seq平台有低背景、高敏感性,但是带有生物学重复的试验设计对于将RNA丰度变化推广到取样群体中仍是 至关重要的。一般,RNA-seq试验设计,包括分组、随机化和重复都已经深入讨论过。
为了解释生物学变异性,为SAGE数据而开发的方法最近被用于RNA-seq数据。两者的主要差异在于数据集的规模。为了解释生物学变异性,使用负二项分 布作为Poisson分布的自然推广,要估计一个额外的散度参数。一些基于负二项分布的计数数据差异表达分析变体涌现出来,包括普通散度模型,用加权似然 率共享所有基因的信息,均值方差关系的经验估计和使用等价类的经验Bayes实现。Poisson模型的包含散度的扩展也已提出,如广义Poisson分 布或两步Poisson模型(在取决于数据中散度证据的两个模式下检验差异表达)。一些实时转录本发现和定量或可变亚型分析的工具也执行差异表达分析。但 是值得注意的是这些方法要么用Poisson分布要么用Fisher 精确检验,显然都不能处理上面讨论的生物学变异。
当前的很多计数数据差异表达分析策略中有很多都限于简单实验设计,如成对或多组比较。据我们所知,还没有针对更复杂设计的分析的通用方法提出来,如配对样
本会时间过程实验,在RNA-seq数据的语境下。缺少这样的方法,研究者就把他们的计数数据转换成有合适工具的连续数据。一般线性模型提供了对上述计数
数据的逻辑推广,而也需要开发更聪明的策略来共享所有基因的信息;目前软件工具提供了这些方法。进一步,上面讨论的方法主要目的在于汇总已有注释的表达水
平。以无目标方式检测差异表达的方法近来也提出来了,如极大均值差异检验。
系统生物学:超越基因列表
在很多情形中,建立差异表达基因列表并非分析的最终步骤;可以通过寻找基因集的表达变化获得对试验系统的深入生物学见解。很多聚焦于基因集检验、网络推断 和知识库的工具为分析微阵列数据集的差异表达基因而设计出来。然而,RNA-seq受到微阵列数据所没有的一些偏倚所影响。例如,基因长度偏倚是RNA- seq数据的一个问题,其中越长的基因有越高的计数。这导致了对长的高表达基因来说,差异表达检测有更高的检定力。这些偏倚极大地影响下游分析结果,如 GO富集。为了能进行基因集分析,Bullard等建议通过除以基因长度的平方根来修改差异表达t-statictic以最小化差异表达中长度偏倚的影 响。另一方面,GOseq特别为RNA-seq数据开发的工具,可以把长度或总计数偏倚合并到基因集检验中。随着对RNA-seq数据偏倚的认识深化,结 合了这种认识的系统生物学工具对于提取出生物学见解是至关重要的。
对于集成RNA-seq数据结果和其他生物学数据源已建立更完整的基因调控图,有着广泛的理解。例如,RNA-seq与基因分型数据结合以鉴定解释个体间
基因表达变异的遗传基因座(表达数量性状位点,eQTLs)。而且,整合表达数据和转录因子结合、RNA干扰、组蛋白修饰以及DNA甲基化信息具有更好理
解各种调控机制的潜力。这种整合性分析的一些报道近来也出现了。例如,Lister等突出了基因体中CG和非CG甲基化水平与RNA-seq表达水平的显
著差异。类似地,测序数据集的组合正开始提供单等位基因与表达、组蛋白修饰和DNA甲基化之间关联性的见解。
Outlook
本评论中,我们列出了处理RNA-seq短reads以进行样本间差异表达分析的主要步骤。简言之,过程就是,映射并汇总短reads序列,然后样本间归 一化并执行差异表达统计检验。进一步的生物学见解可以通过寻找基因集内表达变化模式和整合RNA-seq数据和其它来源的数据来获得。
尽管这个流程的很多部分都是扩展研究的焦点,但仍有些领域存在进一步细化的可能。目前,很少有工作在研究那种汇总度量是最适合寻找样本间差异表达基因的。 为了进行更复杂试验设计的分析,还有扩展现有差异表达检测统计方法的余地。而且,现有的很多方法的相对优点应在进一步的研究中经受考验,依照其分析各种研 究设计的灵活性,其在大大小小的研究中的性能,对测序深度的依赖和强加的假设(如均值方差关系)的准确性。进一步,尽管有很多用RNA-seq进行可变剪 接检测的例子,但是仍有必要扩展当前方法以在生物学变异占主导时检测基因亚型偏好的差异,也许使用上述基于计数的方法。
由于产生短reads的实验协议之间有本质不同,正式比较RNA-seq平台以及很多数据分析方法的相对优点将是很重要的。这样的研究会揭示平台特异的差 异表达分析的好处并会促进更好的数据整合。该领域仍然相对年轻,我们希望在未来有更多的RNA-seq数据分析新方法和工具涌现出来。
{nextpage}
RNA-Seq技术的优缺点
一直以来,研究人员都很有兴趣了解细胞在各种不同状态下的基因表达差异,并开发出多种方法,来不断提高灵敏度和增加通量。基因表达芯片是近年来较多采用的方法,但它如今却碰上了一个强劲对手——RNA-Seq.
RNA-Seq可进行全基因组水平的基因表达差异研究,具有定量更准确、可重复性更高、检测范围更广、分析更可靠等特点。除了分析基因表达水平,RNA-Seq还能发现新的转录本、SNP、剪接变体,并提供等位基因特异的基因表达。
RNA-Seq的动态范围更广,且假阳性可能更小,这意味着RNA-Seq的数据重复性应当比芯片要高。RNA-Seq能够检测样品中的所有RNA,这对 于鉴定细胞的新颖转录本来说是个优点,但同时缺点在于,它检测了总的RNA,而细胞中很大一部分RNA都来自核糖体和线粒体。这限制了其他RNA的读取数 量以及这些RNA表达水平的准确性。因此,polyA RNA选择和核糖体RNA去除等方法被开发出来,以便解决这个问题。
然而,这些分离方法有可能会引入潜在误差,影响实验结果。因此,第三代测序公司Helicos BioSciences的研究人员对操作方法修改后可能发生哪些差异进行了研究,文章发表在最近一期的PloS ONE上。
他们认为,对于研究人员来说,必须了解以下几点:技术差异如何影响结果的质量和可解释性,操作方法如何引入潜在误差,RNA的来源如何影响转录检测,以及所有这些差异如何影响得到的结论。
Helicos BioSciences公司的研究人员使用了多个人RNA样品,来评估RNA片段化、RNA分离、cDNA合成,单个以及多个标签计算。尽管采用 polyA RNA选择的操作方法得到了最多的非核糖体读取,并能够最精确测定编码转录本,但研究人员发现这种方法只能检测人细胞中的一部分非核糖体RNA.
polyA RNA排除了数千个注释的转录本以及更多未注释的转录本,使得转录组查看不完全。而核糖体去除的RNA则提供了一个更经济的方法,来生成完整的转录组覆盖。与多个标签相比,利用单个标签的表达测定具有评估基因表达和检测短RNA的优势。
这项工作有望帮助研究人员在分析基因表达时选择适当的产品。参考文献:
Raz T, Kapranov P, Lipson D, Letovsky S, Milos PM, et al. (2011) Protocol Dependence of Sequencing-Based Gene Expression Measurements.PLoS ONE 6(5): e19287.
doi:10.1371/journal.pone.0019287
{nextpage}
RNA-seq实验的几点经验
作者:高山
随着illumina推出myseq等更便宜的设备,RNA-seq在国内使用越来越多
但是由于国内的垄断等原因,很多公司对客户进行误导
自己基于多个项目和大量数据处理的经验,给大家几点建议,仅供参考
1. 一定要坚定跟着illumina,尽管其他几款设备也有很大进步,但是差距很是不小的;
2. 100bp单向测序足够了,不用paired end为好,后者经常被用了宰客;
3. 组装还是trinity最好,华大那个更快
4. 组装前要对测序的数据做严格的质量控制,去污染(primer、病毒、rRNA)
5. 建议使用strand specific技术
因为RNA-seq制备方法必须反转录成 cDNA,因此丢失了转录体序列的方向性,
为了知道转录本来自具体哪条DNA,人们发明了strand specific。
strand-specific RNA-seq library的制备方法太多了,但是dUTP还是最成熟最可靠的
我们实验室有个protocal又快又便宜,你们可以参考一下
High-Throughput Illumina Strand-Specific RNA Sequencing Library
Preparation
Silin Zhong等
RNA-seq & tools (常用工具和数据库)
http://woldlab.caltech.edu/rnaseq/
http://rna-seqblog.com/data-analysis/rna-seq-data-analysis-tools/
http://rna-seqblog.com/data-analysis/beginner-rna-seq-data-analysis-pipeline/
http://hannonlab.cshl.edu/fastx_toolkit/galaxy.html#fasta_collapser
http://www.ebi.ac.uk/Tools/rcloud/
http://rna-seqblog.com/data-analysis/splicing-junction/new-splice-junction-mapping-tool-mapsplice/
http://rna-seqblog.com/publications/goseq-gene-ontology-go-analysis-on-rna-seq-data/
http://seqanswers.com/forums/showthread.php?t=6148
http://bioinformatics.oxfordjournals.org/content/early/2011/01/13/bioinformatics.btr012.full.pdf
splice RNA-Seq
http://www.web-books.com/MoBio/Free/Ch5A4.htm
http://seqanswers.com/forums/showthread.php?t=3754
Alternative splicing detection using RNA-seq
http://seqanswers.com/forums/showthread.php?t=7811
The GATK can be used with any BAM file for analysis. The CountLoci and CountReads walkers are really for QC. The Pileup and DepthOfCoverage walkers we actually use for analysis, and may be informative for RNA-seq data analysis. But we do not have any specific analysis tools for RNA-seq data, and this is not a development priority for us. Perhaps another member of the community is writing GATK-based tools for RNA-seq analysis, though, and will chime in.
http://en.wikipedia.org/wiki/RNA-Seq
转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP研究),非编码区域功能研究(Non-coding RNA研究、microRNA前体研究等),基因表达水平研究以及全新转录本发现。
转录组(transcriptom)广义上指某一生理条件下,细胞内所有转录产物的 集合;狭义上指所有mRNA的集合。蛋白质是行使细胞功能的主要承担者,蛋白质组是细胞功能和状态的最直接描述,而由于目前蛋白质实验技术的限制,转录组 成为研究基因表达的主要手段。转录组是连接基因组遗传信息与生物功能的蛋白质组的必然纽带,转录水平的调控是目前研究最多的,也是生物体最重要的调控方 式。转录组即特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。转录组研究是基因功能及结构研究的基础和出发点,通 过新一代高通量测序,能够全面快速地获得某一物种特定组织或器官在某一状态下的几乎所有转录本及基因序列,已广泛应用于基础研究、临床诊断和药物研发等领 域。转录组测序可以得到特定条件下所有mRNA转录本的丰度信息,从而发现新的转录本和可变剪接体。应用领域: 1. 非编码区域功能研究:Non-coding RNA研究、microRNA前体研究等 2. 转录本结构研究:UTR鉴定、Intron边界鉴定、可变剪切研究、Start codon鉴定等 3. 基因转录水平研究 4. 全新转录区域研究 基于 Illumina 高通量测序平台的转录组测序技术使能够在单核苷酸水平对任意物种的整体转录活动进行检测,在分析转录本的结构和表达水平的同时,还能发现未知转录本和稀有 转录本,精确地识别可变剪切位点以及cSNP(编码序列单核苷酸多态性),提供最全面的转录组信息。相对于传统的芯片杂交平台,转录组测序无需预先针对已 知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强 大工具。
http://www.nature.com/nmeth/journal/v7/n11/abs/nmeth.1517.html
http://hi.baidu.com/insidi/blog/item/93f74216cdb98243f2de3226.html
http://rna-seqblog.com/data-analysis/expression-tools/rseqtools-a-modular-framework-to-analyze-rna-seq-data-using-compact-anonymized-data-summaries/
http://rna-seqblog.com/
================================
tools
A reasonably thorough table of next-gen-seq software available in the commercial and public domain
Integrated solutions
* CLCbio Genomics Workbench - de novo and reference assembly of Sanger, Roche FLX, Illumina, Helicos, and SOLiD data. Commercial next-gen-seq software that extends the CLCbio Main Workbench software. Includes SNP detection, CHiP-seq, browser and other features. Commercial. Windows, Mac OS X and Linux.
* Galaxy - Galaxy = interactive and reproducible genomics. A job webportal.
* Genomatix - Integrated Solutions for Next Generation Sequencing data analysis.
* JMP Genomics - Next gen visualization and statistics tool from SAS. They are working with NCGR to refine this tool and produce others.
* NextGENe - de novo and reference assembly of Illumina, SOLiD and Roche FLX data. Uses a novel Condensation Assembly Tool approach where reads are joined via "anchors" into mini-contigs before assembly. Includes SNP detection, CHiP-seq, browser and other features. Commercial. Win or MacOS.
* SeqMan Genome Analyser - Software for Next Generation sequence assembly of Illumina, Roche FLX and Sanger data integrating with Lasergene Sequence Analysis software for additional analysis and visualization capabilities. Can use a hybrid templated/de novo approach. Commercial. Win or Mac OS X.
* SHORE - SHORE, for Short Read, is a mapping and analysis pipeline for short DNA sequences produced on a Illumina Genome Analyzer. A suite created by the 1001 Genomes project. Source for POSIX.
* SlimSearch - Fledgling commercial product.
Align/Assemble to a reference
* BFAST - Blat-like Fast Accurate Search Tool. Written by Nils Homer, Stanley F. Nelson and Barry Merriman at UCLA.
* Bowtie - Ultrafast, memory-efficient short read aligner. It aligns short DNA sequences (reads) to the human genome at a rate of 25 million reads per hour on a typical workstation with 2 gigabytes of memory. Uses a Burrows-Wheeler-Transformed (BWT) index. Link to discussion thread here. Written by Ben Langmead and Cole Trapnell. Linux, Windows, and Mac OS X.
* BWA - Heng Lee's BWT Alignment program - a progression from Maq. BWA is a fast light-weighted tool that aligns short sequences to a sequence database, such as the human reference genome. By default, BWA finds an alignment within edit distance 2 to the query sequence. C++ source.
* ELAND - Efficient Large-Scale Alignment of Nucleotide Databases. Whole genome alignments to a reference genome. Written by Illumina author Anthony J. Cox for the Solexa 1G machine.
* Exonerate - Various forms of pairwise alignment (including Smith-Waterman-Gotoh) of DNA/protein against a reference. Authors are Guy St C Slater and Ewan Birney from EMBL. C for POSIX.
* GenomeMapper - GenomeMapper is a short read mapping tool designed for accurate read alignments. It quickly aligns millions of reads either with ungapped or gapped alignments. A tool created by the 1001 Genomes project. Source for POSIX.
* GMAP - GMAP (Genomic Mapping and Alignment Program) for mRNA and EST Sequences. Developed by Thomas Wu and Colin Watanabe at Genentec. C/Perl for Unix.
* gnumap - The Genomic Next-generation Universal MAPper (gnumap) is a program designed to accurately map sequence data obtained from next-generation sequencing machines (specifically that of Solexa/Illumina) back to a genome of any size. It seeks to align reads from nonunique repeats using statistics. From authors at Brigham Young University. C source/Unix.
* MAQ - Mapping and Assembly with Qualities (renamed from MAPASS2). Particularly designed for Illumina with preliminary functions to handle ABI SOLiD data. Written by Heng Li from the Sanger Centre. Features extensive supporting tools for DIP/SNP detection, etc. C++ source
* MOSAIK - MOSAIK produces gapped alignments using the Smith-Waterman algorithm. Features a number of support tools. Support for Roche FLX, Illumina, SOLiD, and Helicos. Written by Michael Str?mberg at Boston College. Win/Linux/MacOSX
* MrFAST and MrsFAST - mrFAST & mrsFAST are designed to map short reads generated with the Illumina platform to reference genome assemblies; in a fast and memory-efficient manner. Robust to INDELs and MrsFAST has a bisulphite mode. Authors are from the University of Washington. C as source.
* MUMmer - MUMmer is a modular system for the rapid whole genome alignment of finished or draft sequence. Released as a package providing an efficient suffix tree library, seed-and-extend alignment, SNP detection, repeat detection, and visualization tools. Version 3.0 was developed by Stefan Kurtz, Adam Phillippy, Arthur L Delcher, Michael Smoot, Martin Shumway, Corina Antonescu and Steven L Salzberg - most of whom are at The Institute for Genomic Research in Maryland, USA. POSIX OS required.
* Novocraft - Tools for reference alignment of paired-end and single-end Illumina reads. Uses a Needleman-Wunsch algorithm. Can support Bis-Seq. Commercial. Available free for evaluation, educational use and for use on open not-for-profit projects. Requires Linux or Mac OS X.
* PASS - It supports Illumina, SOLiD and Roche-FLX data formats and allows the user to modulate very finely the sensitivity of the alignments. Spaced seed intial filter, then NW dynamic algorithm to a SW(like) local alignment. Authors are from CRIBI in Italy. Win/Linux.
* RMAP - Assembles 20 - 64 bp Illumina reads to a FASTA reference genome. By Andrew D. Smith and Zhenyu Xuan at CSHL. (published in BMC Bioinformatics). POSIX OS required.
* SeqMap - Supports up to 5 or more bp mismatches/INDELs. Highly tunable. Written by Hui Jiang from the Wong lab at Stanford. Builds available for most OS's.
* SHRiMP - Assembles to a reference sequence. Developed with Applied Biosystem's colourspace genomic representation in mind. Authors are Michael Brudno and Stephen Rumble at the University of Toronto. POSIX.
* Slider- An application for the Illumina Sequence Analyzer output that uses the probability files instead of the sequence files as an input for alignment to a reference sequence or a set of reference sequences. Authors are from BCGSC. Paper is here.
* SOAP - SOAP (Short Oligonucleotide Alignment Program). A program for efficient gapped and ungapped alignment of short oligonucleotides onto reference sequences. The updated version uses a BWT. Can call SNPs and INDELs. Author is Ruiqiang Li at the Beijing Genomics Institute. C++, POSIX.
* SSAHA - SSAHA (Sequence Search and Alignment by Hashing Algorithm) is a tool for rapidly finding near exact matches in DNA or protein databases using a hash table. Developed at the Sanger Centre by Zemin Ning, Anthony Cox and James Mullikin. C++ for Linux/Alpha.
* SOCS - Aligns SOLiD data. SOCS is built on an iterative variation of the Rabin-Karp string search algorithm, which uses hashing to reduce the set of possible matches, drastically increasing search speed. Authors are Ondov B, Varadarajan A, Passalacqua KD and Bergman NH.
* SWIFT - The SWIFT suit is a software collection for fast index-based sequence comparison. It contains: SWIFT — fast local alignment search, guaranteeing to find epsilon-matches between two sequences. SWIFT BALSAM — a very fast program to find semiglobal non-gapped alignments based on k-mer seeds. Authors are Kim Rasmussen (SWIFT) and Wolfgang Gerlach (SWIFT BALSAM)
* SXOligoSearch - SXOligoSearch is a commercial platform offered by the Malaysian basedSynamatix. Will align Illumina reads against a range of Refseq RNA or NCBI genome builds for a number of organisms. Web Portal. OS independent.
* Vmatch - A versatile software tool for efficiently solving large scale sequence matching tasks. Vmatch subsumes the software tool REPuter, but is much more general, with a very flexible user interface, and improved space and time requirements. Essentially a large string matching toolbox. POSIX.
* Zoom - ZOOM (Zillions Of Oligos Mapped) is designed to map millions of short reads, emerged by next-generation sequencing technology, back to the reference genomes, and carry out post-ana
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#RNA-Seq#
42