NEJM:英国“破译发育障碍”项目揭示五千余例罕见病的遗传原因
2023-05-16 测序中国 测序中国 发表于上海

在该文章中,研究团队描述了DDD研究多年来开发的分析策略。
导读
近年来,基因组测序在识别罕见单基因疾病的分子机制方面取得了非凡的进展,并越来越多地应用于多种疾病的诊断。最早将大规模基因组研究与个体患者反馈相结合的研究之一是英国破译发育障碍(Deciphering Developmental Disorders, DDD)研究项目。该项目招募了13500多个家庭,获得了外显子组测序和微阵列数据,并与200多名临床医生记录的丰富临床表型相补充。
近日,该项目团队在国际顶级期刊The New England Journal of Medicine发表了题为“Genomic Diagnosis of Rare Pediatric Disease in the United Kingdom and Ireland”的文章。在该文章中,研究团队描述了DDD研究多年来开发的分析策略。通过该项目,大约5500例严重发育障碍患者了解了其疾病相关遗传因素。平均而言,每个父母-后代三人组中有1.0个候选变异,每个单例先证者有2.5个变异。通过使用临床和计算方法进行变异分类,约41%的先证者(13,449人中有5502人)得到诊断,其中76%的个体携带致病性新生突变。另外22%的先证者(13449人中的2997人)在与单基因发育障碍密切相关的基因中存在意义不明变异。

文章发表在NEJM
该研究纳入的所有家庭都有患有严重发育障碍的儿童,但这些儿童并没有被明确诊断,且很可能是由单一基因变化引起。截至目前,该团队已经为大约5500名儿童提供了基因诊断,这些诊断来自800多个不同的基因,包括该研究之前发现的60种新疾病相关分子。其中,大约四分之三的疾病是由自发突变引起的,而不是遗传自父母任何一方。
文章第一作者、英国埃克塞特大学基因组医学教授Caroline F. Wright表示:“对患有罕见疾病的家庭来说,得到正确的诊断绝对至关重要,这些家庭大约每17人中就有1人患有罕见疾病。这些疾病大多是遗传性的,使用基因组测序技术进行诊断可能对临床管理和患者的生活质量产生巨大影响。目前,我们与数百名临床医生和科学家,以及数千名患者合作,试图找到这些答案。希望通过分享我们的发现,未来会有更多家庭更快地得到答案。”
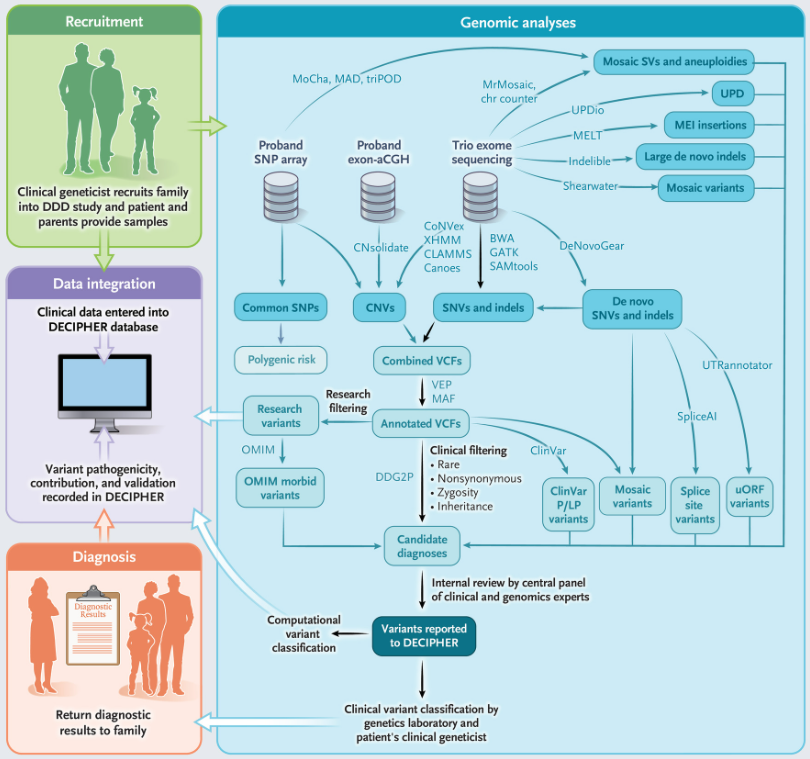
图1. 研究设计及流程示意图,来源:NEJM
共有13449个先证者(9859名为父母-后代家族)被纳入该研究,72%的先证者是其家族中唯一受影响的成员。研究团队进行了三种独立的基因组分析:完整家庭和单例先证者DNA样本的外显子测序,先证者DNA外显子微阵列比较基因组杂交(aCGH),以及先证者DNA全基因组单核苷酸多态性(SNP)基因分型,并使用多种不同算法来检测和注释序列和结构变异。
分析显示,先证者中发现了19,285个潜在的致病序列和结构变异,并通过多达6轮的迭代再分析和批量报告反馈给临床医生,这些分析涉及18种不同变异检测算法。大多数变异是通过DDG2P数据库确定的,DDG2P数据库是一个由1840个发育障碍相关基因组成的临床数据库,该数据库以每年约100个基因的速度更新。DDG2P中包括60个该研究新发现的发育障碍相关基因。

图2. 研究发现的变异。来源:NEJM
该研究报告的变异中有44%是在2014年第一轮报告后被添加到DDG2P数据库中的基因。大多数报告的变异为单核苷酸变异(SNV)和使用外显子组测序数据检测到的小片段插入或缺失(Indel,71%是蛋白质改变,19%是蛋白质截断,3%是非编码变异);结构变异是通过微阵列和外显子组测序分析的组合确定的(6%是拷贝数变异,1%是其他结构变异)。平均而言,每个父母-后代三人组中有1.0个候选变异,每个单例先证者有2.5个变异。
研究发现,先证者的从头变异和来自父母的嵌合遗传变异(即合子后亲本基因中的新生变异)提供了最高的诊断率,79%的变异被临床归类为致病或可能致病。相比之下,32%的常染色体隐性变异、23%的X染色体母系遗传变异和11%的常染色体显性变异被临床分类为致病或可能致病。
此外,临床与该研究预测变异的致病性和良性分类的一致性很高,共有4425个一致变异。对应于研究团队使用的混合方法敏感性为99.5%,特异性为85.0%,阳性预测值为96.5%,阴性预测值为97.9%。149个不一致变异(3%)是由于假阳性变异检测、不正确的临床分类或不适当的ACMG-AMP或ACGS标准分配导致。
根据预测变异致病性分类与临床之间的一致性,研究团队估计13,449个先证者中至少有3347人(25%)得到了诊断;对于接受预测诊断的先证者,这一比例增加到4363(32%);对于通过临床诊断的先证者,这一比例增加到4484(33%);对于临床或预测诊断的先证者,这一比例增加到5502(41%)(图3)。
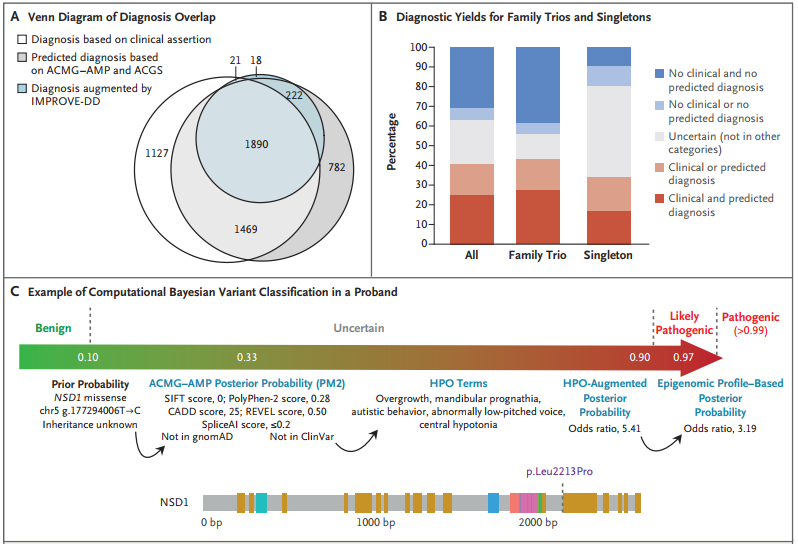
图3. DDD项目的诊断流程,来源:NEJM
在通过临床获得诊断的4484名先证者中,有3599人属于父母-后代家庭,其中2750人(76%)携带致病性新生变异。在4484个先证者中,561人(13%)接受了部分诊断,121个(3%)接受了两种或两种以上不同的遗传诊断,可能导致复合表型。13449个先证者中,携带意义不明变异的先证者为2997人(22%),其中0.7%变异的预测贝叶斯后验致病性概率为0.8~0.9。
通过使用混合临床研究模型对大型临床队列进行基因组分析,DDD研究显示了临床专业知识、基因组科学和生物信息学的融合如何能够在标准的、表型驱动的诊断方法失败的案例中推动疾病诊断和发现。
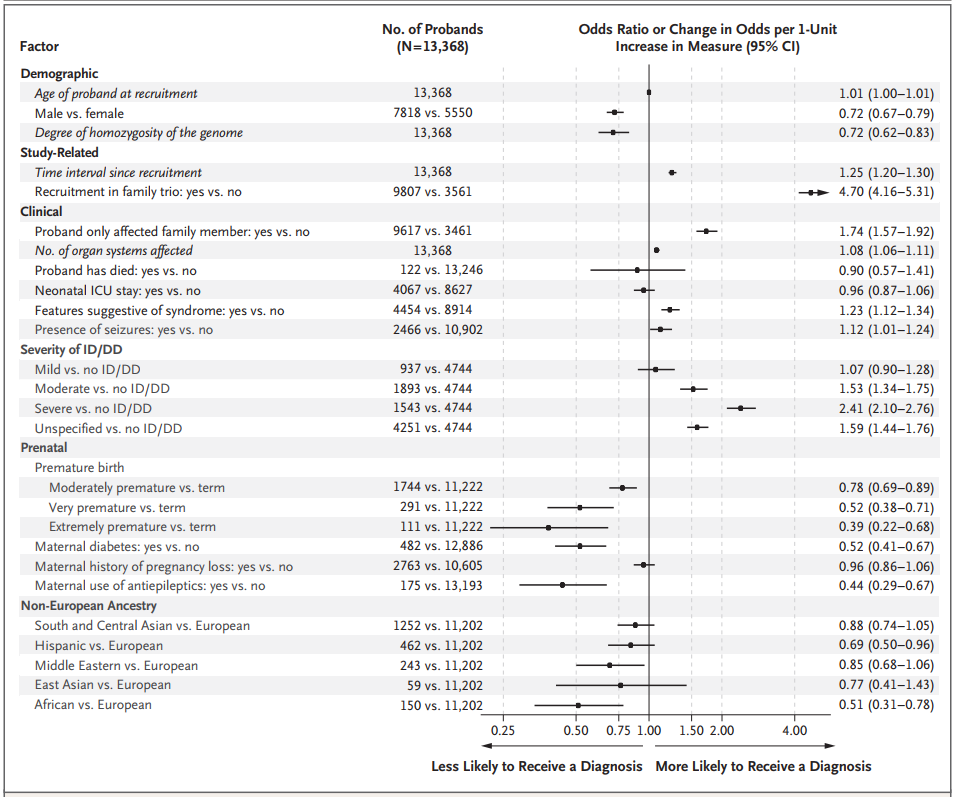
图4. 影响诊断的因素汇总,来源:NEJM
文章资深作者、剑桥大学临床基因组学教授Helen V. Firth强调了信息平台在支持招募患者和将诊断结果反馈给临床团队方面的重要性。他表示:“在这项研究的核心嵌入一个强大的信息学平台,促进了同参与该项目的家庭、临床医生和科学家的合作,并在诊断成功、发现并最终治疗罕见基因组疾病的新机制方面发挥了至关重要的作用。”
NHS的基因组医学服务部门以及为可能患有急性单基因疾病的儿童提供快速基因组测序服务的部门,现在正在使用类似的方法来诊断罕见疾病个体,该服务可以在十天内为正在或面临重症监护的婴儿和儿童提供基因诊断。
英国卫生部长Will Quince表示:“我们正在创建世界上最先进的基因组医疗保健系统,这项研究是英国国民健康医疗改革的又一步。使用这样的尖端高科技方法,有可能更好地了解和更准确地诊断罕见遗传疾病,使儿童能够更快地获得治疗,并有可能改善疾病对他们生活的影响。”
DDD研究项目已经成功造福数千个未明确诊断的受发育障碍影响的家庭。分析数据表明,基因组驱动方法与详细的表型分析相结合,可以提高诊断精度。该项目的数据分析也强调了使用多样化变异检测方法,结合严格的变异过滤规则和重复的、迭代的变异分析和分类,可以从现有数据中做出新的诊断。
参考资料:
1. Genomic Diagnosis Rare Pediatric Disease in the United Kingdom and Ireland, New England Journal of Medicine (2023).
https://www.nejm.org/doi/10.1056/NEJMoa2209046
2.Study sheds light on causes of rare genetic diseases in 5,500 people
https://medicalxpress.com/news/2023-04-rare-genetic-diseases-people.htm
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言