Cell Reports:南方医科大学朱浩团队开发深度学习算法DeepFavored,揭示适应性突变与疾病易感性关联
2022-09-27 测序中国 测序中国

人类和黑猩猩之间的基因差异仅为1%,但就是这1%的差异将人类与黑猩猩区分开来。因此,人类基因组中的许多微小变化决定了人类特有的特征。基因组的单核苷酸多态性(SNP)使人类能够适应不同的环境和生活方式,
近日,中国南方医科大学朱浩教授团队在Cell Reports期刊发表了题为“Uncovering the extensive trade-off between adaptive evolution and disease susceptibility”的研究文章。研究团队开发了一种名为DeepFavored的深度学习算法,该算法通过对已有的全基因组关联研究(GWAS)数据集进行统计测试,可以区分有利突变与“搭便车”突变。研究团队在三个不同的人群中验证了该工具,并确定了基因组进化的协调性:适应特定环境的突变也使人们更容易患上某些疾病,或者携带搭便车突变。 文章发表在Cell Reports上
由于搭便车突变与有利突变非常相似,任何方法都很难同时准确地区分两者以及普通突变(图1)。为了避免错误识别,研究团队通过构建深度学习网络,利用不同数据组成训练网络,最后使用训练网络从统计检测的选择信号中鉴定有利突变,最终开发了DeepFavored深度学习算法。此外,研究人员系统地比较DeepFavored与iSAFE、SWIF(r)的性能,这两种方法是识别有利突变的最新方法。结果显示,DeepFavored的性能优于这两种算法,且更加稳定。
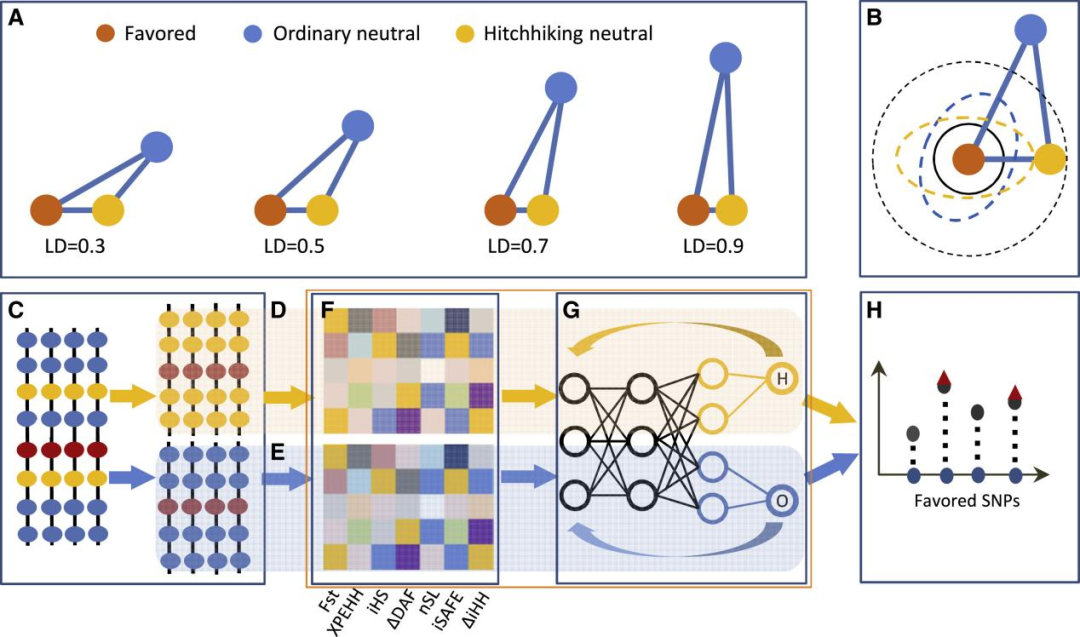
图1. DeepFavored算法设计原理、输入和输出。
随后,研究团队利用真实世界的GWAS数据检测了东亚(CHB)、欧洲(CEU)、非洲(YRI)三个人群的基因组突变,并比较了DeepFavored、iSAFE和SWIF(r)的性能。该研究重点分析了与饮食相关的基因等位基因,包括与代谢或味觉以及免疫相关的基因,并假设上述三个群体需要适应与病原体和食物供应相关的不同压力。
研究人员分析了CEU、CHB和YRI中的1091个PopHumanScan基因区域,这些区域是识别有利突变的良好候选区域。最终,DeepFavored、iSAFE和SWIF(r)分别在CEU、CHB和YRI中鉴定了1,013(454、287、272)、1,219(560、487、172)和789(350、334、104)个有利突变。此外,以上三种方法共同识别了55个有利突变,研究团队利用这55个有利突变作为测试数据进一步评估了DeepFavored的性能。结果表明,由DeepFavored识别的有利突变应该是合理的。
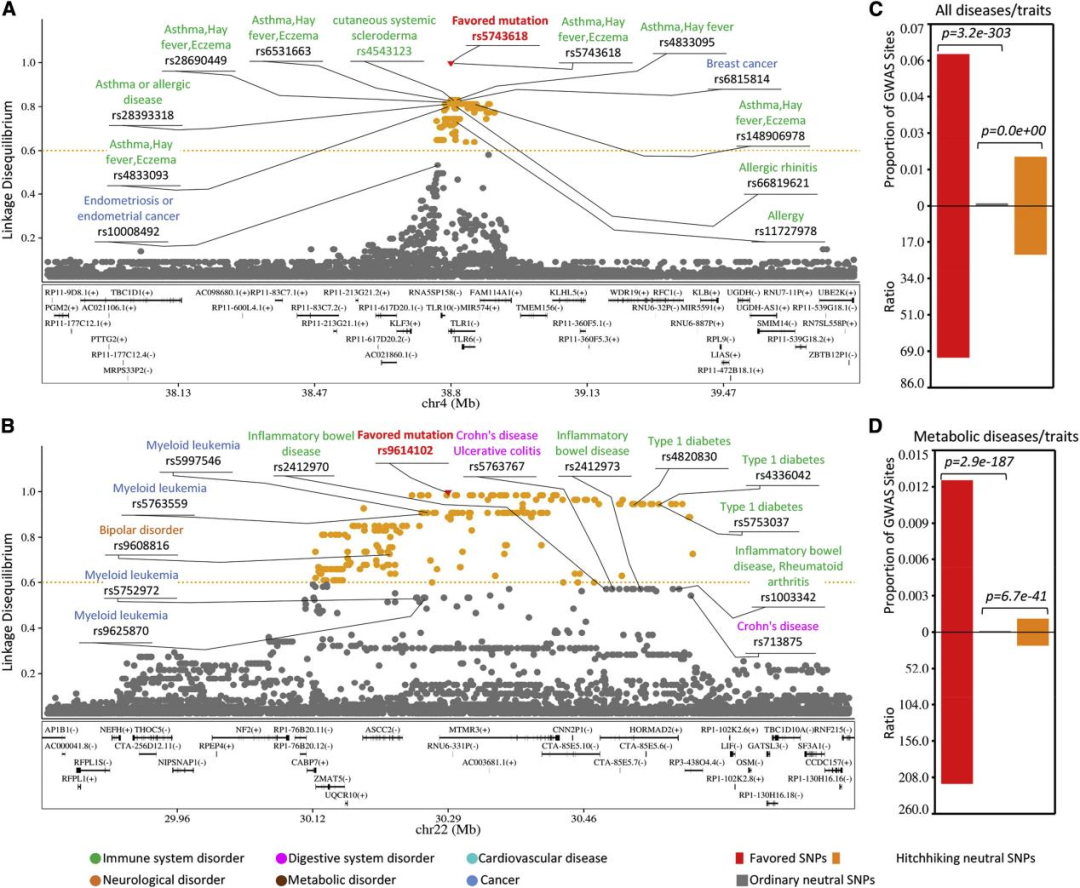
图2. 有利突变和搭便车突变在GWAS位点富集。
在确定了三个人类群体的有利突变后,研究团队综合分析了有利/搭便车突变与GWAS位点之间的相关性。通过计算有利突变、搭便车突变和普通突变的GWAS位点的比例,发现有利突变和搭车突变中GWAS位点的比例明显高于普通突变。
为了检验这种基因突变协调性的潜在相关人群特异性和疾病特异性,研究团队分析了DeepFavored在CEU、CHB和YRI中识别的454、287和272个有利突变,以及附近的搭便车突变是否富含神经、代谢和免疫相关疾病/性状位点。结果表明,有利突变和搭便车突变富含GWAS位点,并且这种富集表现出人群和疾病特异性特征(图3)。

图3.不同突变的GWAS位点。
环境变化、病原体进化以及生活方式改变驱动着基因组的适应性进化。基因组区域的许多变化可能会产生有害的搭便车突变。当新的外部变化发生时,有利突变可能会变得不适应,甚至变得与疾病相关。越来越多的研究强调了适应性进化和疾病易感性之间的协调性,但其规模和细节还不清楚。研究团队开发的深度学习算法DeepFavored能够系识别有利突变,为适应性进化和疾病易感性之间的广泛协调性分析提供了证据。此外,人群特异性有利突变和疾病相关突变的联合分析,可以为精准医疗提供有价值的数据和线索。
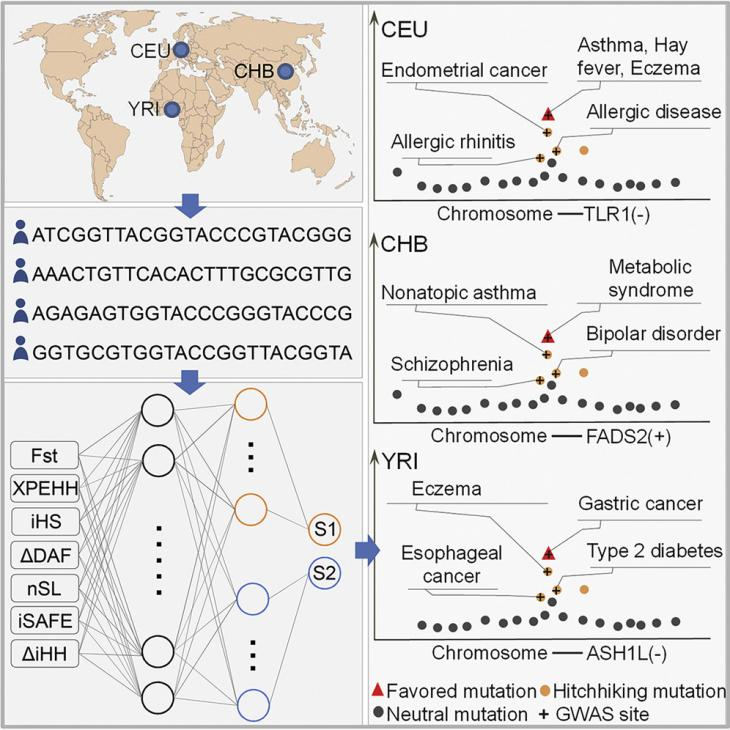
图4. 研究概要。
参考资料:
1.Ji Tang, Maosheng Huang, Sha He, et al. Uncovering the extensive trade-off between adaptive evolution and disease susceptibility. Cell Reports, 2022. DOI:https://doi.org/10.1016/j.celrep.2022.111351
https://www.cell.com/cell-reports/fulltext/S2211-1247(22)01179-2#secsectitle0080
2.Historic Adaptations May Now Make Us Susceptible to Disease
https://www.the-scientist.com/news-opinion/historic-adaptations-may-now-make-us-susceptible-to-disease-70506
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言