罕见病研究无人区——中国学者在先天性巨大黑痣基础研究领域取得新进展
2022-09-26 MedSci原创 MedSci原创

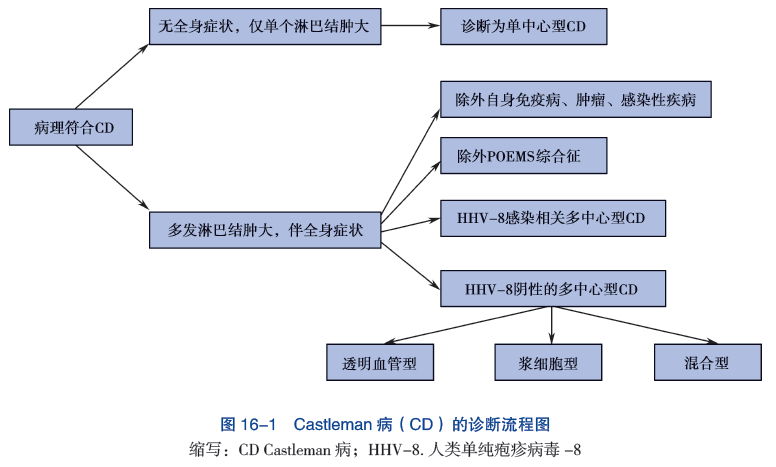
目前,对先天性巨大黑痣的发生、恶变机制尚未完全清楚,治疗也仅停留在手术切除层面,对于累及皮肤面积过大的患者,无手术治疗的可能。
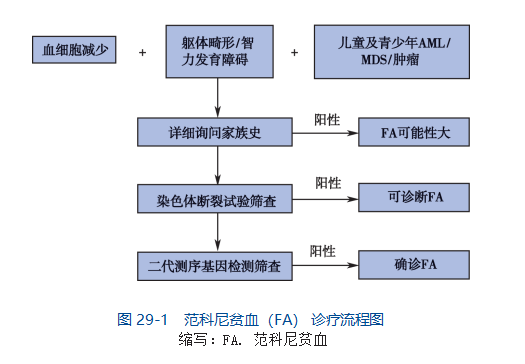
先天性巨大黑痣(giant congenital melanocytic nevi, GCMN)又称黑毛痣、兽皮痣,指出生即有的,累及大面积皮肤的黑痣,在新生儿中发病率约为1/20000。该病最大可累及超过80%的患儿体表面积,表面可粗糙被毛,严重影响患儿容貌,同时相关研究报道其终身恶变几率可高达5%-10%,另有约3%-12%的GCMN患者伴有中枢神经系统累及,称为神经皮肤黑变病,病变可导致患儿脑积水,癫痫发作以及神经功能损害。目前对GCMN治疗手段有限,对于较小面积的病灶,尚能手术切除,对累及全身大部面积的患者尚无相关治疗手段。在基础研究领域,受限于体内外研究模型平台的缺乏,目前对GCMN的基础研究开展极为困难。
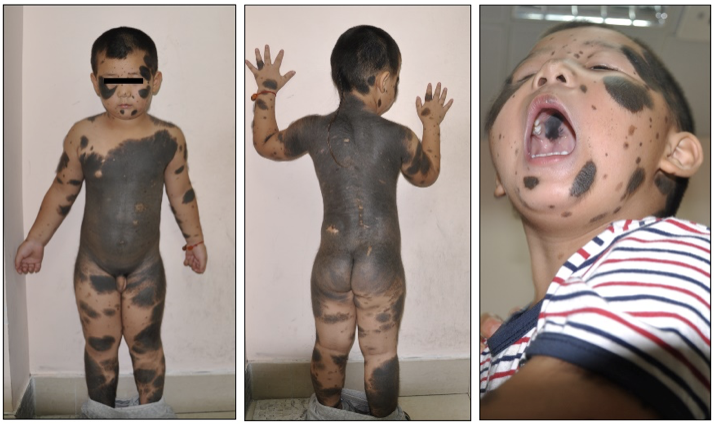
近日国际色素细胞学会联合会会刊《色素细胞及黑色素瘤研究》(Pigment Cell & Melanoma Research, JCR Q1)发表了上海第九人民医院整复外科李青峰教授团队的最新研究成果——成功构建首株先天性巨大黑痣永生化细胞株(Establishment and characterization of an immortalized human giant congenital melanocytic nevi cell line)。据悉,以往无可长期培养的先天性巨大黑痣良性细胞模型,这极大限制了该疾病的相关基础研究,为弥补这一空白,整复外科博士后余庆雄在导师李青峰教授的指导下,从一例GCMN患者手术切除的巨痣皮肤组织中分离培养巨痣细胞,并经过SV40T转染进行永生化,建立国际上首株永生化先天性巨大黑痣细胞系GNC-9H-01,经鉴定,该细胞系能稳定传代,并伴显著色素合成。经基因测序发现,该细胞株携带杂合NRASQ61K基因突变。裸鼠种植实验显示该细胞系能在小鼠皮下成活形成种植瘤。目前该细胞系保藏于中国典型细胞保藏中心,并已进行了发明专利申请。
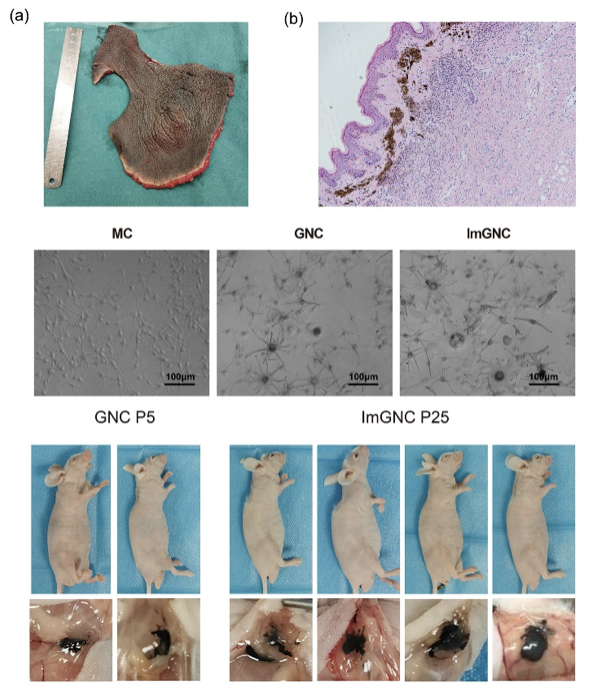
动物模型也是疾病研究中的重要工具,李青峰教授团队也致力于GCMN小鼠模型的构建。基于前期对中国GCMN患者群体的大样本量全外测序研究,余庆雄博士后发现中国患者群体中NRASQ61K突变基因发生率是NRASQ61R突变的2.3倍。因此团队选择了构建黑色素细胞选择性表达NRASQ61K突变基因的小鼠模型(Dpp3-Cre/NRASQ61K),该模型小鼠成功表现出与人先GCMN类似的表型。目前,九院整复外科是继瑞士苏黎世大学、哈佛大学麻省总院皮肤生物中心之外第三个,也是国内首个成功构建先天性巨大黑痣模型小鼠的研究中心。余庆雄博士后介绍,相对于国外的两种模型小鼠,九院自主构建的模型小鼠方法稳定可靠,背景一致性高,繁育方案简单,且更贴近中国GCMN患者突变特征。据悉,该模型小鼠已申请国家发明专利。

目前对先天性巨大黑痣的发生、恶变机制尚未完全清楚,治疗也仅停留在手术切除层面,对于累及皮肤面积过大的患者,无手术治疗的可能。九院整复外科作为全国最大的整形外科中心之一,每年接诊大量的先天性巨大黑痣患者,而临床治疗手段的进展亟需基础研究领域的突破,目前构建的GCMN体内外研究模型平台极大拓展了相关基础研究的开展。据悉,目前李青峰教授团队在已构建的GCMN体内外模型平台的基础上,联合瑞金医院国家转化医学中心(上海)药物筛选平台,积极推进GCMN靶向治疗药物筛选及验证,未来有望开发自主知识产权的GCMN靶向治疗药物,为广大的先天性巨大黑痣患者带来福音。
原始出处
Yu, Qingxiong, et al. "Establishment and characterization of an immortalized human giant congenital melanocytic nevi cell line." Pigment Cell & Melanoma Research 35.3 (2022): 356-368.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言