FDA上半年批准新药达25款,创历史新高
2020-07-18 药明康德,医药魔方 药明康德,医药魔方

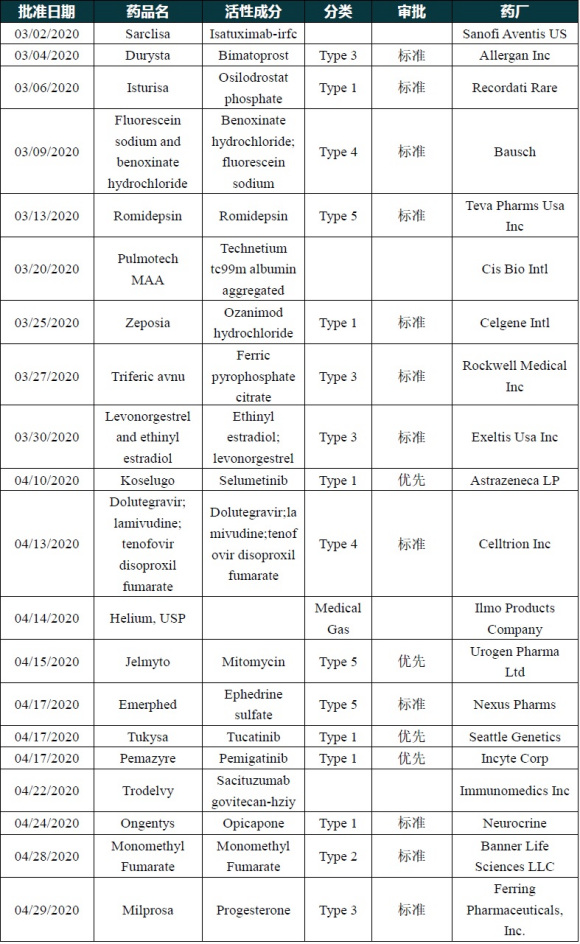
2020年的上半年,美国FDA的药物评估与研究中心(CEDR)已经批准了25款创新药。
2020年的上半年,美国FDA的药物评估与研究中心(CEDR)已经批准了25款创新药。包括19个新分子实体和6个新生物制品。从获批个数来看,上半年虽然新冠疫情全球流行,但FDA批准的新药数量较去年同期的14款增幅不小。也是近10年来的多个“新高”。
下图内容至6月28日,在6月30日FDA又批准了triheptanoin,共25款。

美国FDA在2020年上半年批准新药一览(数据来源:FDA官网)
FDA上半年新药批准数目创10年来新高
当新冠疫情席卷全球之时,FDA能否在新冠疫情的压力下,按时完成对创新药的审评和批准工作,是业界关注的焦点之一。毕竟新冠疫情不但给FDA的工作人员增加了工作负担,也为多种审评流程和专家咨询会的召开设置了障碍。不过,从FDA今年上半年的新药批准数量来看,新冠疫情尚未显著影响到新药获批的速度。今年上半年FDA批准的新药数量在近10年中排名第一,甚至超过了获批总数创纪录的2018年的上半年表现。
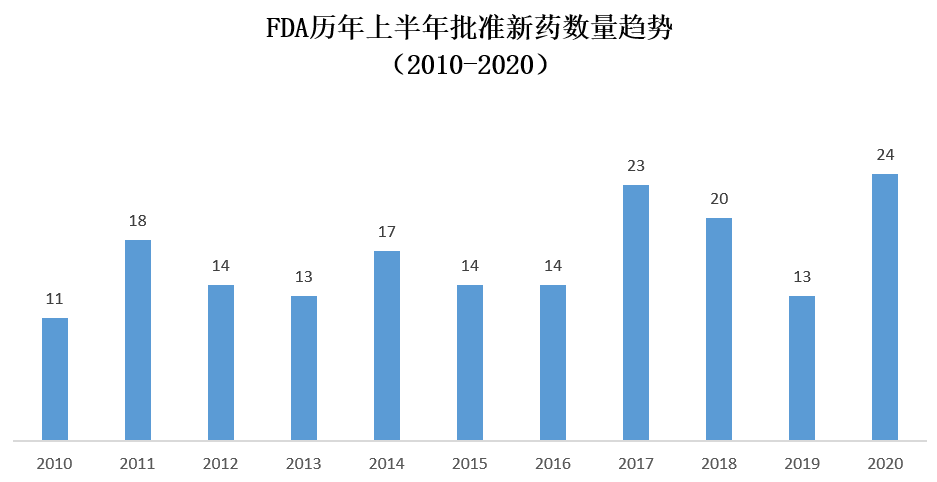
美国FDA历年上半年批准新药数目(数据来源:FDA官网)
诚然,影响新药获批数目的原因是多方面的,涉及到新药申请递交的时间,不同加速通道的使用,以及FDA审批流程现代化等多种因素。而且,FDA官员也表示,虽然上半年的新药批准进度令人满意,但是如果新冠疫情持续发展,也不能保证以后的新药获批进度仍然能够得到保持。但是,FDA在新冠疫情中的表现,表明了它仍然将把创新疗法带给急需的患者作为工作的首要目标之一。
孤儿药获批比例创10年来新高
近10年来,获得FDA批准的孤儿药在获批创新疗法中的比例逐步上升。今年上半年,获得孤儿药资格的创新疗法获得批准的数目为14个,占上半年获批新药58.3%。从比例上看,达到了10年来占比的新高,比2018年的峰值(57.6%)还要高出一线。
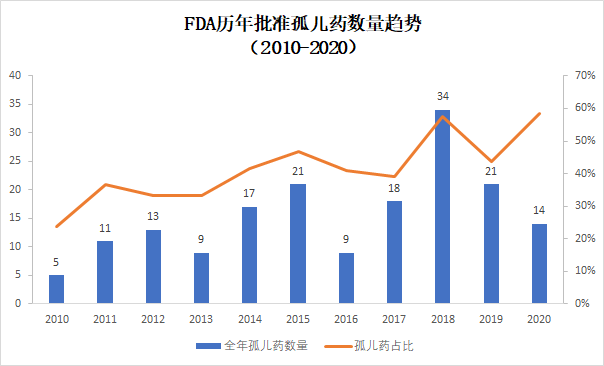
▲2010-2020年孤儿药获批数量和占比(数据来源:FDA官网,药明康德内容团队制图,2010-2019年数据为全年数据,2020年为上半年数据)
针对罕见病的孤儿药开发不但得益于FDA颁发的多种监管措施的激励,也体现了医药行业对罕见病疗法开发的重视。今年上半年获批的孤儿药中很多是针对患者数目稀少的罕见病。例如首款治疗1型神经纤维瘤病的药物疗法Koselugo(Selumetinib)。
另一方面,随着对癌症分子生物学特征的进一步了解,研发人员能够根据肿瘤携带的特定突变开发具有针对性的精准疗法。将癌症患者根据分子生物学特征进行细分,使治疗患者的数目也达到了获得孤儿药资格的标准,这也增加了孤儿药获批的数目。今年获批的首款新药,Blueprint Medicines公司开发的Ayvakit(Avapritinib)就是一个例子。
精准疗法获批占比创10年来新高
如上面所说,分子生物学技术的发展,让研究人员能够更详细地发现驱动癌症发展的基因变异,并且针对这些基因变异开发精准疗法。这些疗法由于特异性非常强,往往在具有良好疗效的同时,降低了疗法的毒副作用。今年上半年,FDA总计批准了4款精准疗法(定义为需要使用FDA批准的伴随诊断确定患者携带特定基因变异的疗法),占获批创新疗法的17%,达到近10年来占比值的高峰。上面提到的Ayvakit,和首款获得FDA批准的MET抑制剂Tabrecta(Capmatinib)都是这一药物开发策略的范例。
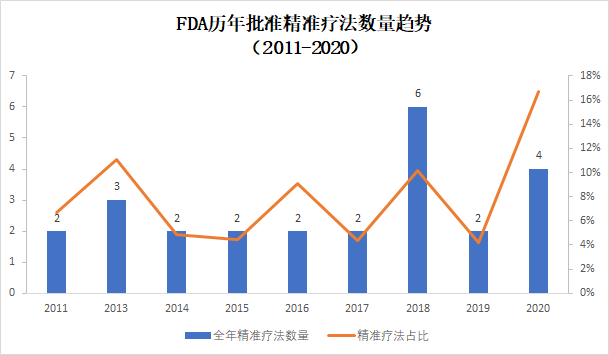
2011-2020年精准疗法获批数量和占比(数据来源:FDA官网,2011-2019年数据为全年数据,2020年为上半年数据)
从历史数据中可以看出,精准疗法获批占比在过去十年中没有太多变化。然而,随着下一代测序技术的进一步普及,以及液体活检技术的进一步完善,个体化治疗的策略正在被医药行业普遍采纳。我们希望今年上半年多款精准疗法的批准,成为精准疗法爆发的先兆。
抗体偶联药物的光明未来
今年上半年,FDA批准了Immunomedics公司开发的抗体偶联药物Trodelvy(sacituzumab govitecan-hziy)上市,治疗三阴性乳腺癌患者。这也是近一年半以来,FDA批准的第4款抗体偶联药物,占FDA总共批准的抗体偶联药物的50%。
抗体偶联药物(ADC)的开发并非一帆风顺,虽然这一概念早在上世纪70年代就被提出,但是到2000年,辉瑞的Mylotarg才成为FDA批准的首款ADC。然而,因为毒性原因它在2010年撤市。开发成功的ADC需要特异性高的抗体,毒性强的细胞毒性分子,以及稳固的连接子(linker),在保证足够的药物能够达到肿瘤细胞内部,产生杀伤活性的同时,降低对健康组织的伤害。过去的几十年里,业界投入大量资源和精力改进构建ADC的技术。
可喜的是,近年来多款ADC的获批显示,对ADC构建技术的改进有效地提高了ADC药物的获益/风险比。新一代的ADC不但携带全新的细胞毒性分子(例如去年底获批的Enhertu),而且在控制药物抗体比例(ADR)和连接子稳定性方面也做出了长足的进步。使用非天然氨基酸,研究人员能够精确控制偶联在抗体上的细胞毒性分子的数目和偶联位点,让ADC的安全性控制更为精准。
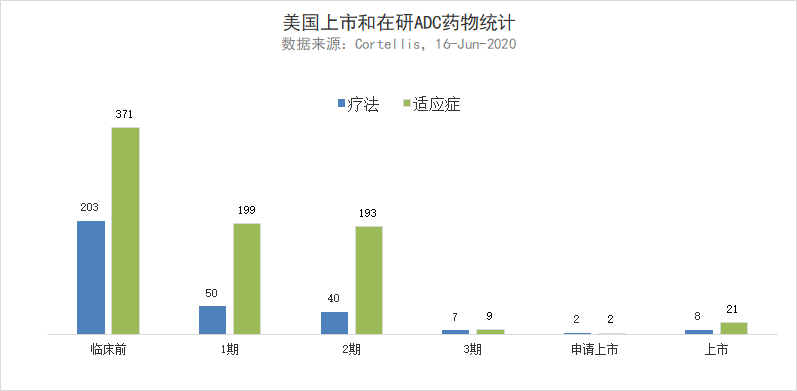
美国上市和在研ADC药物统计(数据来源:Cortellis,截至2020年6月16日)
对ADC研发管线的统计也表明,目前在临床1/2期阶段,有大量在研ADC疗法在用于治疗几百种适应症。ADC技术的进步也让研究人员不再局限于将细胞毒性分子偶联在抗体上,新一代ADC能够将免疫调节因子等其它药物类型偶联在抗体上,治疗癌症以外的其它疾病。
我们期待随着ADC技术的不断完善,这一治疗模式能够发挥出它最大的潜力。
1.avapritinib
商品名:Ayvakit
公司:Blueprint Medicines
1月9日,FDA批准Blueprint Medicines公司的Ayvakit (avapritinib)上市,用于治疗携带血小板源性生长因子受体 α(PDGFRA)基因18外显子突变的不可手术或转移性成人胃肠道间质瘤(GIST)患者,其中也包括D842V 位点突变这种最常见的18外显子跳跃突变类型。
Avapritinib是一种口服的、具有高活性高选择性的KIT及PDGFRα抑制剂。具有KIT及PDGFRα突变(包括KIT D816V、PDGFRα D842V 和KIT 外显子17突变)的多种疾病对标准疗法响应很差,缺乏有效的治疗方式,avapritinib针对以上突变在临床前均表现了很好的活性。
GIST是一类起源于胃肠道间叶组织的肿瘤,占消化道间叶肿瘤的大部分。GIST 可以发生于消化道的任何部位,以胃和小肠最为常见,分子机制是编码酪氨酸激酶受体蛋白基因 KIT(CD117) 或血小板源性生长因子受体 α(PDGFRα) 基因激活突变所致。大约6%的新确诊患者携带PDGFA外显子18突变。
2.teprotumumab-trbw
商品名:Tepezza
公司:Horizon Therapeutics
1月21日,FDA批准Horizon Therapeutics的Tepezza(teprotumumab-trbw)上市,用于治疗甲状腺眼病。这是FDA批准的首个甲状腺眼病药物。
TED是一种常发生于甲亢患者身上的自身免疫性疾病,临床通常表现为眼球突出、复视、视力模糊和面部畸形等。该疾病主要是由于患者自身抗体激活了眼框内胰岛素样生长因子1受体(IGF-1R)所导致。
Teprotumumab是一种靶向IGF-1R的单克隆抗体,III期OPTIC临床研究表明,接受Teprotumumab治疗的患者较安慰剂能显著减轻患者眼球突出症(82.9% vs 9.5%),到达主要终点。另外,接受该药治疗的患者在减轻复视、改善生活质量等方面均有效果,到达次要终点。安全性方面,Teprotumumab治疗组发生的大多数不良反应为轻度至中度,并且可以控制。
3.tazemetostat
商品名:Tazverik
公司:Epizyme
1月24日,FDA批准Epizyme公司的Tazverik(tazemetostat)上市,用于治疗无法完全切除的转移性/局部晚期上皮样肉瘤成人患者以及16岁以上的儿科患者。
上皮样肉瘤是一种罕见的软组织肉瘤,好发于20~40岁青壮年,男性较为多见。目前,主要的治疗方案有手术切除、化疗或放疗。Tazemetostat是FDA批准的首个EZH2抑制剂,也是FDA批准的首个上皮样肉瘤治疗药物。
一项II期临床研究结果表明,接受Tazemetostat治疗的患者ORR达到15%,其中CR为1.6%,PR为13%。另外,在所有产生应答的患者中,67%的患者持续反应时间达到6个月或更长时间。
安全性方面,Tazemetostat治疗组中有37%的患者发生严重不良反应。其中≥3%的患者发生的严重不良反应主要为出血、胸腔积液、皮肤感染、呼吸困难、疼痛和呼吸窘迫。1例患者(2%)患者因情绪变化的不良反应而永久停用该药。
4.lactitol
商品名:Pizensy
公司:Braintree实验室
2月12日,FDA批准Braintree实验室开发的Pizensy(lactitol)上市,用于治疗慢性特发性便秘(CIC)成人患者。
CIC是一种以排便困难、排便次数不频繁、腹痛腹胀为主要特征的疾病,在美国,大约有3500万人受到这一疾病的困扰。目前,临床上主要以包括泻药在内的药物进行治疗,这类药物虽然能一定程度缓解病人症状,但同时也带来了药物依赖。
Pizensy是一种口服乳糖醇疗法,属于渗透性泻药,它能够促进水分进入肠道,进而达到通便作用。一项随机、双盲、安慰剂对照的多中心临床试验结果表明,Pizensy治疗组较安慰剂组患者在一周内达到3次完全自发排便次数(CSBMs)或者较基线比至少增加1次CSBMs的患者比例分别为25%和13%,到达主要终点。
安全性方面,常见的不良反应为上呼吸道感染、腹胀、腹泻、血肌酐磷酸激酶升高、腹胀和血压升高。
5.bempedoic acid
商品名:Nexletol
公司:Esperion
2月21日,FDA批准Esperion公司的Nexletol(bempedoic acid)片剂上市,辅助饮食和最大耐受剂量他汀用于治疗成人杂合子型家族性高胆固醇血症(HeFH)患者,或者用于需要进一步降低LDL-C的动脉粥样硬化性心血管疾病(ASCVD)患者。Nexletol是自2002年以来批准的首个每日1次口服非他汀类降LDL-C药物。
bempedoic acid是一种人工合成的二羧酸衍生物,属于first in class的ATP-柠檬酸裂解酶(ACL)抑制剂,通过抑制肝脏内胆固醇的生物合成和上调低密度脂蛋白受体(LDL-R)来降低LDL-C。
FDA批准Nexletol主要基于一系列在超过3000例患者中开展的全球关键III期项目的结果。对其中4项合计超过2600例患者的临床试验聚合分析结果显示,bempedoic acid联合中等剂量或最高耐受剂量他汀可以使安慰剂校正后的LDL-C进一步降低18%,bempedoic acid联合低剂量他汀或不使用他汀,可使安慰剂校正后的LDL-C进一步降低21%~28%。
在III期临床研究中,bempedoic acid的耐受性良好,常见不良事件的发生率与安慰剂组相似,最常见(≥2%)且高于安慰剂组的不良事件主要包括上呼吸道感染、肌肉痉挛、高尿酸血症、背痛、腹痛或轻度不适、支气管炎、贫血、肝酶升高等。不良事件严重程度方面,bempedoic acid组大多数都是轻至中度,与安慰剂组类似。
6.eptinezumab-jjmr
商品名:Vyepti
公司:灵北制药
2月21日,FDA批准灵北制药的Vyepti(eptinezumab-jjmr)上市,用于成人偏头痛的预防治疗。这是FDA批准的首个预防偏头痛的静脉注射药物。
Vyepti是灵北斥资19.5亿美元收购Alder公司所获得一款降钙素基因相关肽(CGRP)药物,CGRP被认为是一种与偏头痛相关的重要神经肽。
Vyepti的疗效和安全性在两项随机、双盲、安慰剂对照的临床研究中得到证实。其中,PROMISE-1研究主要针对发作性偏头痛患者,PROMISE-2研究主要针对慢性偏头痛患者。结果表明,Vyepti在这两项研究中均到达了降低患者1-3个月内平均每月偏头痛天数(MMD)的主要终点。另外在一项涉及2076例患者的安全性研究表明,Vyepti最常见的不良反应为鼻咽炎和超敏反应。
7.amisulpride
商品名:Barhemsys
公司:Acacia Pharma
2月27日,FDA批准Acacia Pharma公司的Barhemsys(amisulpride)上市,作为单药或联合其它疗法,治疗和预防患者术后恶心和呕吐症状(PONV)。这是首个获批的对于先前预防失败患者的术后恶心呕吐补救疗法。
PONV是外科手术的常见并发症,大约30%的手术患者和80%的高危患者都会发生这一症状。PONV与麻醉气体或镇痛药物的使用有关,在妇科、腹部、乳房、眼睛和耳朵的手术后,尤其在持续一个小时及以上时间的手术后,更为常见。部分患者调查显示,PONV被列为手术并发症中最严重的症状之一,甚至超过疼痛。
Barhemsys是一种选择性多巴胺D2和D3受体拮抗剂。Barhemsys的疗效和安全性在4项III期临床研究中得到证实。其中,一项涉及标准止吐疗法预防失败患者的的临床研究结果表明,Barhemsys治疗组治疗效果显著优于安慰剂组(42% vs 29%)。另一项涉及PONV高风险患者的临床研究结果表明,Barhemsys联合另一项止吐药的治疗效果显著由于安慰剂联合该止吐药的效果(58%vs 47%)。
8.rimegepant
商品名:Nurtec ODT
公司:Biohaven
2月27日,FDA批准Biohaven公司的Nurtec(rimegepant)口腔崩解片上市,用于成人急性偏头痛的治疗。
Rimegepant是一种口腔崩解片(ODT),可在无水(或仅有少量水存在)的条件下于口腔中快速崩解,随吞咽动作进入消化道,在口腔内无粘膜吸收。与普通制剂相比,ODT有服用方便、吸收快、生物利用度高、对消化道黏膜刺激性小等优点,受到广泛关注。
FDA对rimegepant的批准基于一项关键的III期临床试验(Study 303)和一项长期开放标签安全性研究(Study 201)的结果。
在Study 303研究中,与安慰剂相比,在服用rimegepant后2小时,达到无疼痛和大多数烦人症状(MBS)的共同主要终点方面具有一致的统计学显著差异,rimegepant在缓解疼痛(将中度或重度疼痛减轻为无痛或轻度疼痛)和在1小时内恢复正常功能方面也显示出统计上的优势。对于大多数患者而言,疼痛自由、疼痛缓解、恢复正常功能以及摆脱MBS的益处持续了48小时。重要的是,只需1剂rimegepant就可以看到这些获益。服用rimegepant的患者中有86%在给药后24小时内不需要急救药物(例如NSAIDS,对乙酰氨基酚)。
Study 201研究评估了长期多次服用rimegepant的安全性和耐受性。该研究评估了1798例偏头痛发作的患者,这些患者根据需要服用75mg的rimegepant,每天最多服用1剂。该研究包括1131例暴露于rimegepant至少6个月和863例暴露于至少1年的患者,所有这些患者平均每月至少发生2次偏头痛发作,但该研究尚未确定rimegepant在30天内出现15次偏头痛发作患者身上的安全性。
在临床研究中,少于1%的受试者发生呼吸困难和严重皮疹的超敏反应,包括给药后延迟的严重超敏反应。
9.isatuximab
商品名:Sarclisa
公司:赛诺菲
3月2日,FDA批准赛诺菲的CD38单抗药物Sarclisa (isatuximab-irfc)上市,用于联合泊马度胺和地塞米松治疗既往至少接受过2线以上疗法(包括来那度胺和一种蛋白酶体抑制剂)的复发性难治性多发性骨髓瘤(MM)成人患者。
isatuximab通过靶向多发性骨髓瘤细胞上CD38受体的特定表位促进肿瘤细胞程序性死亡,是继强生Darzalex(达雷妥尤单抗)之后全球第2款获FDA批准的CD38抗体药物。2019年,Darzalex销售额达到29.98亿美元。
此次批准是基于ICARIA-MM临床试验,结果显示,Sarclisa联合泊马度胺+地塞米松治疗组较泊马度胺+地塞米松治疗组患者的疾病进展和死亡风险降低了40%(HR 0.596),二者的中位无进展生存期(PFS)分别为11.53 和 6.47个月;同时,Sarclisa联合泊马度胺+地塞米松治疗组较泊马度胺+地塞米松治疗组患者的总体缓解率显著提高(60.4%vs. 35.3%)。
安全性方面,Sarclisa联合治疗组最常见的不良反应为中性粒细胞减少症(96%)、输液相关反应(39%)、肺炎(31%)、上呼吸道感染(57%)和腹泻(26%);超过5%的患者发生严重不良反应,包括肺炎(25.3%)和发热性中性粒细胞减少(12.3%);7%的患者因不良反应(3-4级)而停用Sarclisa联合治疗,3%的患者因输液相关反应而停止治疗。
10.osilodrostat
商品名:Isturisa
公司:诺华
3月7日,FDA批准诺华的Isturisa(osilodrostat)上市,用于治疗库欣综合征。这是FDA批准的首个口服11‐β‐羟化酶抑制剂。
库欣综合征,又称皮质醇增多症,是一种由于多种原因引起的肾上腺皮质长期分泌过多糖皮质激素所产生的临床症候群。库欣综合征是一种罕见疾病(发病率为1~2/100万),患者通常表现为满月脸、多血质外貌、向心性肥胖、痤疮、紫纹、高血压、继发性糖尿病和骨质疏松等。该类疾病高发年龄为20~50岁,女性发病率为男性的3倍。
Isturisa是一种皮质醇合成抑制剂,可抑制11-β羟化酶的产生而起到治疗作用,11-β羟化酶是负责肾上腺皮质醇合成最终步骤的酶。研究结果表明,Isturisa可使库欣综合征患者皮质水平正常化,并改善其他临床特征。在III期LINC‐3研究中,接受Isturisa治疗的患者在停药8周后保持正常平均尿游离皮质醇的比例显著高于安慰剂(86% vs 29%)。安全性方面,常见的不良反应为肾上腺功能不全、疲劳、恶心、头痛和水肿。
11.ozanimod
商品名:Zeposia
公司:BMS
3月26日,FDA批准BMS的Zeposia (ozanimod) 上市,用于治疗成人复发型多发性硬化症(RMS),包括临床孤立综合征、复发缓解型多发性硬化症(RRMS)、以及活动性继发进展型多发性硬化症(SPMS)。
MS是一种人体免疫系统异常攻击大脑、脊髓、视神经的神经细胞髓鞘而引起的慢性疾病,表现为肌肉虚弱、疲劳、视物困难,最终导致残疾。全球大约有230万例MS患者,其中85%在初次确诊时属于复发缓解型MS(RRMS),15%属于原发进展型MS(PPMS)。RRMS患者表现为周期性的疾病复发与缓解,随着疾病的进展,最终会恶化成继发性进展型多发性硬化症(SPMS)。PPMS患者则表现为症状持续恶化,没有明显的缓解期。MS目前尚无法完全治愈。
Ozanimod是鞘氨醇1-磷酸受体(S1PR)调节剂,对淋巴细胞表面的S1PR1和S1PR5均有较高的亲和力。当Ozanimod与受体的结合发生后,可以阻止淋巴细胞离开淋巴结进入中枢神经系统,起到抗炎作用。此外,siponimod也能进入中枢神经系统,直接与少突胶质细胞和星形胶质细胞上的S1PR结合,促进髓鞘再生和防止炎症。
12.selumetinib
商品名:Koselugo
公司:阿斯利康/默沙东
4月10日,FDA批准阿斯利康/默沙东的Koselugo(selumetinib)上市,用于2岁及以上儿童和青少年的神经纤维瘤病I型(NF1)患者,这些患者伴有症状性和/或进展性、无法手术的丛状神经纤维瘤(PN)。这是FDA批准的首个用于治疗NF1的药物。
神经纤维瘤病(NF)是一种良性的周围及中枢神经系统疾病,属于常染色体显性遗传病。根据其临床表现和基因定位位点不同,主要分为神经纤维瘤病1型( NF1)和神经纤维瘤病2型(NF2)。
据估计,NF1发病率大约为1/3000~4000个新生儿。大约20~50%的NF1患者可能为丛状神经纤维瘤,这种肿瘤往往不能完全手术切除,复发风险高。NF1患者可能会伴有许多并发症,如学习困难、视力障碍、脊椎扭曲和弯曲、高血压和癫痫。NF1还会增加一个人患其他癌症的风险,包括恶性脑瘤和白血病等。
Selumetinib是一款MEK 1/2抑制剂,MEK是RAS/MAPK信号通路中的关键蛋白激酶,而NF1的发病正是由于NF1基因突变扰乱了RAS/MAPK信号通路所导致的肿瘤生长。该药曾获得FDA授予的孤儿药资格和突破性治疗法称号。目前,这款药物已经在中国获批临床。
13.pemigatinib
商品名:Pemazyre
公司:Incyte公司
4月17日,FDA加速批准Incyte公司Pemazyre(pemigatinib),用于治疗既往接受过治疗的携带FGFR2融合或重排的局部晚期或转移性胆管癌患者,批准时间比预定的5月30日审批日期提前了一个半月,这也是FDA批准的首个胆管癌靶向疗法。
pemigatinib是一种选择性的成纤维细胞生长因子受体(FGFR)抑制剂,对FGFR1、FGFR2、FGFR3均有抑制作用。FGFRs在肿瘤细胞增殖、生存、迁移、新生血管形成中发挥着重要作用。FGFRs基因的融合、重排、易位和扩增与多种癌症的发生和进展密切相关。FDA曾授予pemigatinib用于接受过治疗的携带FGFR2基因易位的晚期/转移性或不可手术切除的胆管癌患者突破性疗法资格,以及治疗胆管癌的孤儿药资格。
14.tucatinib
商品名:Tukysa
公司:Seattle Genetics
4月17日,FDA批准Seattle Genetics公司Tukysa(tucatinib)上市,联合化疗(曲妥珠单抗和卡培他滨)用于治疗无法通过手术切除并且之前已经接受过一种以上疗法的HER2阳性晚期乳腺癌患者。这是FDA在“ORBIS计划”下批准的首个新分子实体药物,比预定审批期限提前了4个月。
FDA此次批准Tukysa主要基于一项入组612例HER2突变阳性的晚期不可手术切除或转移性乳腺癌患者的临床试研究结果,这些患者既往接受过曲妥珠单抗、帕妥珠单抗和T-DM1治疗,其中48%在入组时已经发生脑转移。研究的主要终点是无进展生存期(PFS),次要终点是脑转移患者的OS和PFS。
结果显示,与安慰剂组+曲妥珠单抗+卡培他滨治疗组相比,Tukysa+曲妥珠单抗+卡培他滨治疗组患者的中位PFS显著提高(7.8 vs 5.6个月),OS显著提高(21.9 vs 17.4个月),脑转移患者的PFS也显著提高(7.6 vs 5.4个月)。
Tukysa治疗组患者最常见的不良反应包括腹泻,手脚灼热或刺痛,恶心,疲劳,肝损伤,呕吐,口腔炎,食欲下降 ,腹痛,头痛,贫血和皮疹等。
15.sacituzumab govitecan-hziy
商品名:Trodelvy
公司:Immunomedics
4月22日,FDA加速批准Immunomedics公司的抗体偶联药物Trodelvy(sacituzumab govitecan-hziy)上市,用于治疗既往接受过至少2种疗法的转移性三阴乳腺癌成人患者。Trodelvy是FDA批准的首个治疗三阴乳腺癌的抗体偶联药物,也是首个获批的靶向人滋养层细胞表面抗原2(Trop-2)的抗体偶联药物。
FDA此次批准是基于一项多中心、单臂、II期临床试验中的客观应答率(ORR)和应答持续时间(DoR)数据。108例既往接受过3线(中位数,范围:2~10)治疗的三阴乳腺癌患者的ORR为33.3%(95% CI:24.6%~43.1%),中位DoR为7.7个月(95% CI:4.9~10.8个月)。
Trodelvy的药品标签中带有黑框警告,提示其有严重中性粒细胞减少和腹泻的风险。II期试验中Trodelvy治疗最常见(≥25%)的不良反应包括恶心、中心粒细胞减少、腹泻、疲劳、贫血、呕吐、脱发、便秘、食欲减退、红疹、腹痛。发生率≥5%的3/4级严重不良反应包括中性粒细胞减少、白细胞计数减少、贫血、低磷血症、腹泻、疲劳、恶心、呕吐。2%的患者因为不良事件终止了治疗,未见治疗相关的死亡病例,未见严重中性粒细胞减少或间质性肺病的病例。
16.opicapone
商品名:Ongentys
公司:Neurocrine Biosciences
4月24日,FDA批准Neurocrine Biosciences公司的Ongentys(opicapone)上市,作为左旋多巴/卡比多巴的辅助疗法,用于治疗处于关闭期的帕金森患者。
帕金森是仅次于阿尔茨海默氏病的美国第二大最常见的神经退行性疾病。据估计,美国帕金森患者大约有100万例,每年新增确诊患者大约为5万例。
opicapone是一种口服选择性COMT抑制剂,能通过抑制分解左旋多巴的COMT酶,进而减少其在血液中的分解,使得更多药物到达患者大脑,最终起到延长疗效和实现患者运动功能的作用。
17.capmatinib
商品名:Tabrecta
公司:诺华
5月6日,FDA批准诺华Tabrecta (capmatinib) 上市,用于治疗局部晚期或转移性MET外显子14跳跃(METEX 14)突变的非小细胞肺癌(NSCLC)患者。FDA同时批准了FoundationOne CDx(F1CDx)作为Tabrecta的伴随诊断产品。
FDA此项批准主要基于一项代号为GEOMETRY mono-1的II期研究数据。该项开放标签、多中心、单臂研究招募了97例经RNA临床分析确证携带MET外显子14跳跃的NSCLC患者,给予capmatinib 400mg直至疾病进展或出现不可耐受毒性,主要终点是总体应答率(ORR),其他终点还包括应答持续期(DOR)。
独立数据监察委员会评估结果显示,capmatinib 针对初治患者的ORR为68%,其中CR 4%,PR 64%,DOR超过12个月的患者比例为47%;针对既往接受过治疗患者的ORR为41%,DOR超过12个月的患者比例为32%。
安全性方面,capmatinib最常见的不良反应为周围水肿(42%)、恶心(33%)、肌酐升高(20%)、呕吐( 19%)、疲劳(14%)、食欲下降(13%)和腹泻(11%)。大部分AE等级为1/2级。
18.selpercatinib
商品名:Retevmo
公司:礼来
5月8日,FDA加速批准礼来Retevmo(selpercatinib,LOXO-292)胶囊上市,用于治疗:1)成人转移性RET融合阳性NSCLC患者,2)需要系统治疗的成人及12岁以上儿科晚期或转移性RET突变阳性甲状腺髓样癌患者,3)放射性碘难治的需要系统治疗的成人及12岁以上儿科晚期或转移性RET融合阳性甲状腺癌患者。
FDA此次批准是基于一项涉及702例RET驱动型癌症患者的I/II期LIBRETTO-001试验的ORR和DoR结果。该研究既纳入了初治患者,也纳入了经治的各种晚期实体肿瘤患者,包括RET融合阳性的NSCLC、RET突变的MTC、RET融合阳性的甲状腺癌以及其它RET改变的实体肿瘤。结果如下:
图片来源:礼来官方微信
在高达50%的RET融合阳性的非小细胞肺癌患者中存在脑转移的情况。在经治的可检测到脑转移的NSCLC患者中,11例患者中有10例观察到颅内病灶的缓解 (CNS ORR),所有10例患者的CNS DoR均≥6个月。
Retevmo最常见的副作用是天冬氨酸转氨酶(AST)升高、丙氨酸转氨酶(ALT)酶升高、血糖升高,白细胞计数减少,白蛋白减少,血钙水平降低,口干, 腹泻,肌酐增加,碱性磷酸酶增加,高血压,疲劳,身体或四肢肿胀,血小板计数低,胆固醇增加,皮疹,便秘和血钠水平降低等。
19.ripretinib
商品名:Qinlock
公司:Deciphera Pharmaceuticals
5月15日,Deciphera Pharmaceuticals公司宣布FDA批准其Qinlock(ripretinib)上市,用于治疗既往接受过伊马替尼、舒尼替尼和瑞戈非尼治疗的晚期胃肠道间质瘤(GIST)患者。
Ripretinib是一种被设计成广谱抑制KIT和PDGFRα突变的研究性酪氨酸激酶开关控制抑制剂,通过独特的双重作用机制,调节激酶开关口袋和激活环。III期INVICTUS研究结果显示,Ripretinib较安慰剂能够改善四线及四线以上GIST患者的无进展生存期(6.3 vs 1.0个月),除此之外,Ripretinib较安慰剂还延长了患者中位总生存期(15.1 vs 6.6个月)。
20.fluoroestrdiolF18
商品名:Cerianna
公司:Zionexa
5月20日,FDA批准Zionexa公司的Cerianna(fluoroestrdiol F18)上市,Cerianna是一种放射学诊断试剂,适用于使用正电子发射断层扫描(PET)成像,作为活检的辅助手段,检测复发/转移性乳腺癌患者的雌激素受体(ER)阳性病变。
21.artesunate
商品名:Artesunate
公司:Amivas
5月26日,FDA批准Amivas公司的Artesunate(artesunate,青蒿琥酯静脉注射剂)上市,用于治疗重症疟疾成人和儿童患者。
青蒿琥酯静脉注射剂的疗效和安全性在1项亚洲开展的随机对照试验(试验1)和1项非洲开展的支持性随机对照试验中(试验1)得到证实。
试验1共入组1461例患者(包括202例年龄不到15岁的儿童患者),试验2共入组5425例年龄小于15岁的重症疟疾患者。这两项研究中,入组患者分别接受青蒿琥酯和对照药物奎宁的治疗。结果显示,青蒿琥酯治疗组患者死亡率显著低于奎宁治疗组。
安全性方面,试验1中患者常见的不良反应为急性肾功能衰竭(需要透析)、血红蛋白尿和黄疸。试验2的安全性与试验1基本相似。
22.flortaucipir F18
商品名:Tauvid
公司:礼来
5月28日,FDA批准礼来的Tauvid(flortaucipirF18)上市,Tauvid是一种放射学诊断试剂,适用于使用正电子发射断层扫描(PET)成像,评估阿尔茨海默病患者大脑中聚集的tau神经纤维缠结(NFTs)的密度和分布。
23.inebilizumab
商品名:Uplizna
公司:Viela Bio公司
6月11日,FDA批准Viela Bio公司的CD19单抗Uplizna(inebilizumab)上市,用于治疗AQP4阳性的成人视神经脊髓炎谱系疾病(NMOSD)患者。豪森药业曾于2019/5/28与Viela Bio公司达成协议,斥资2.2亿美元获得了该药中国权益。
此次批准是基于一项代号为N-Momentum的关键性临床试验结果。该研究共纳入230例NMOSD患者,其中包括213例AQP4阳性患者。结果显示,接受治疗6个月后,89%的AQP4阳性患者疾病没有复发,而安慰剂组这一数值为58%,到达主要终点。另外,接受inebilizumab治疗的患者在关键的次要终点上也显示出显著的统计学差异,包括降低NMOSD相关的住院率。
inebilizumab安全性和耐受性良好,最常见的不良反应为尿路感染(20%)、鼻咽炎(13%)、输液反应(12%)、关节痛(11%)和头痛(10%)。
NMOSD是一种罕见自身免疫疾病,患者体内过度活跃的免疫细胞和自身抗体会攻击视神经和脊髓,导致患者会出现失明、截瘫、感觉丧失、膀胱失调、以及外周疼痛等症状。据估计,80%的NMOSD患者AQP4抗体呈阳性。inebilizumab可以结合B细胞表面的CD19抗原,耗竭CD19+B细胞。
24.lurbinectedin
商品名:Zepzelca
公司:Jazz制药公司/PharmaMar
6月15日,FDA加速批准Jazz制药公司/PharmaMar公司的Zepzelca(lurbinectedin) 上市,用于治疗铂类药物化疗后疾病进展的转移性小细胞肺癌。该药美国市场销售权益归Jazz制药所有,中国开发及商业化权益则归绿叶制药所有。
lurbinectedin是海鞘素衍生物,为RNA聚合酶II的抑制剂,能够与DNA双螺旋结构上的小沟共价结合,抑制RMG1和RMG2,使肿瘤细胞在有丝分裂过程中畸变、凋亡、最终减少细胞增殖。2018年8月,FDA授予lurbinectedin用于治疗小细胞肺癌的孤儿药资格。
25.triheptanoin
商品名:Dojolvi
公司:Ultragenyx公司
6月30日,FDA批准Ultragenyx公司的Dojolvi (triheptanoin)上市,作为卡路里和脂肪酸的来源,治疗通过分子诊断确诊的长链脂肪酸氧化障碍 (LC-FAOD)儿科和成人患者。这是FDA批准的首个LC-FAOD疗法。
LC-FAOD是一种以代谢缺陷为特征的常染色体隐形遗传病,患者由于不能将长链脂肪酸转化为能量,进而导致体内葡萄糖的严重耗尽和并发症,并最终导致住院或死亡。LC-FAOD目前已被纳入美国和部分欧洲国家的新生儿筛查项目中。LC-FAOD的其他治疗选择包括避免禁食和低脂肪/高碳水化合物饮食等。据估计,美国受LC-FAOD影响的患者人数大约为2000-3500例。
Dojolvi是一种高纯度、药用级的奇数碳、中链甘油三酯,该药通过多步骤化学合成,在甘油骨架上添加了3个7碳脂肪酸生成。Dojolvi旨在为LC-FAOD患者提供中链和奇数碳脂肪酸作为能源和代谢物替代品。
详细:首款脂代谢紊乱药物Triheptanoin被FDA批准上市
参考资料:
[1] Novel Drug Approvals for 2020. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2020
[2] How the Next Generation Antibody Drug Conjugates Expands Beyond Cytotoxic Payloads for Cancer Therapy. Retrieved June 28, 2020, from https://www.adcreview.com/articles/how-the-next-generation-antibody-drug-conjugates-expands-beyond-cytotoxic-payloads-for-cancer-therapy/
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#历史#
45
学习
126