肺动脉高压的遗传咨询和检测—国际PAH遗传研究联盟共识
2022-11-04 肺动脉高压研究进展 肺动脉高压研究进展

肺动脉高压是一种罕见的疾病,可由(可能)致病的种系基因组变异引起。除了最普遍的疾病基因,即骨形态发生蛋白受体2外,现在还知道其他几个基因,有些属于不同的功能类别,容易导致PAH的发生。
肺动脉高压(PAH)是一种罕见的疾病,可由(可能)致病的种系基因组变异引起。除了最普遍的疾病基因,即骨形态发生蛋白受体2(BMPR2)外,现在还知道其他几个基因,有些属于不同的功能类别,容易导致PAH的发生。因此,专业和非专业的临床医生和卫生保健专业人员,越来越多地面临着一系列关于PAH患者和/或相关家庭成员的基因测试的需求、方法、益处和风险的问题。该声明提供了一个基于共识的方法来推荐遗传咨询和评估当前疾病基因测试的最佳做法,并且提供了一个框架,以及通过遗传咨询过程向患者和亲属提供的信息类型,并描述了目前已知的疾病致病基因,以便进行分析。将分子基因检测纳入PAH患者管理方案的好处包括:识别被其他诊断方法错误分类的个体,优化表型特征以汇总结果数据,包括在临床试验中,重要的是通过级联筛查,发现健康的因果变体携带者,应向他们提供定期评估。
PAH历史回顾及全球现状
对PAH作为一个独立实体疾病的首次描述是由德国医生Ernst von Romberg作出的。他在1891年的尸检中将肺血管疾病描述为 "肺血管硬化"。自20世纪中期,当右心导管检查(RHC)成为可能后,特发性PAH (IPAH,当时称为原发性肺动脉高压,PPH)被认可。根据法国和苏格兰临床登记研究,PAH的发病率为每年每百万人中有2.5至7.5例,患病率为每百万人中有15至50例。在美国REVEAL登记研究和美国 PAH生物样本中,与其他疾病相关的PAH患者(APAH)构成了最大的 亚人群(分别为50.7%和48.2%),结缔组织疾病占APAH病例的一半,紧随其后的是IPAH。
1954年,Dresdale描述了一种可能的遗传起源,他观察了家族性病例。在20世纪80年代,在14个PPH/PAH家族中描述了一种常染色体显性遗传模式。在90年代末和2000年,PPH1位点被映射到2q31-32号染色体上。2000年,骨形态发生蛋白受体2型(BMPR2)基因的杂合致病性种系变异被发现是造成大多数家族性PAH(FPAH)的原因。这一发现开辟了一个新的时代,突出了PAH的遗传原因。导致与PAH相关的其他几个基因得到鉴定。这一过程中,临床专家将PAH临床分类进行了细化。在2008年第四届世界肺动脉高压研讨会上,遗传性PAH(HPAH)被归类为第1组(PAH)的一个单独的亚类。在最新的PH分类中,这种区分仍然包括 IPAH(1.1组)和HPAH(1.2组)的子类型。所有PAH患者中约有3%被定性为HPAH,约40%为IPAH。
来自多个PAH临床登记研究的数据显示,女性患者占多数;平均70-80%的患者是女性,根据亚组的不同而不同,例如,在与结缔组织疾病相关的PAH中,女性高达90%。然而,在不同的登记资料中有差异,但在老年患者,特别是有吸烟史的患者和青春期前的儿童中,女性占主导地位可能不太明显甚至没有。
PAH的遗传学进展
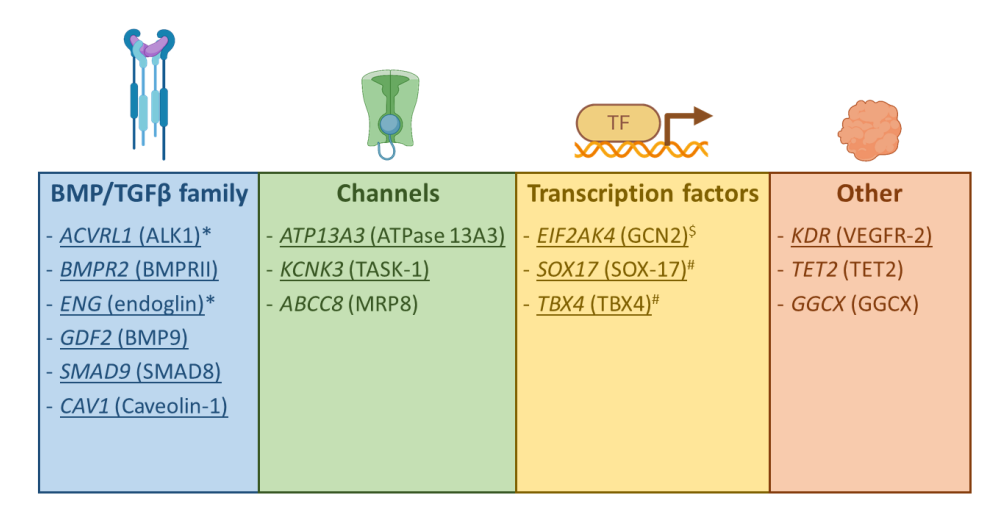
HPAH一词包括PAH的家族性病例和散发性病例,当易感基因中存在潜在的(可能的)致病变体时。约70-87%的家族性PAH和12-20%的IPAH患者中,可以在目前已知的PAH致病基因中找到遗传方面的原因。HPAH最常由BMPR2基因的杂合致病变体引起,该基因编码的是骨形态发生蛋白受体家族的跨膜丝氨酸/苏氨酸激酶(BMPRII)成员,其致病变体通过驱动细胞增殖和阻止细胞凋亡,易导致肺小动脉的狭窄。血管重塑导致肺动脉平滑肌细胞和成纤维细胞的增殖。这可导致新内膜病变和复杂的丛状病变的形成。迄今为止,BMPR2基因中已经发现了800多个不同的、独立的致病变体位置。此外,到2018年,共有17个PAH基因在第六届世界肺动脉高压研讨会上被确认。其中许多基因已被证明属于BMPR2和转化生长因子β(TGF-β)途径或与之相关。
PAH的主要遗传模式是常染色体显性遗传,具有不完全外显率。因此,一个遗传或新产生的(新的)变体就足以导致疾病的发生。然而,并非所有杂合子个体都会发展成PAH。据估计,BMPR2变异体的外显率约为30%,其中42%的女性和14%的男性会患PAH。因此,BMPR2的外显率也有性别差异,女性BMPR2变体携带者的PAH外显率至少是男性变体携带者的两倍。因此,该病可能会 "跳过 "一代,而在下一代再次表现出来。值得注意的是,少数PAH基因也可以是常染色体隐性遗传或半显性作用。已有证据表明,具有双拷贝变体的患者表现出更严重和更早的临床表型。与经典的PAH相比,肺静脉闭塞症和/或肺毛细血管瘤病(PVOD/PCH)的遗传形式的特点是常染色体隐性遗传,原因是真核翻译起始因子2α激酶4(EIF2AK4)基因的双亲致病变体。PVOD患者通常表现为一氧化碳扩散能力(DLCO)降低和计算机断层扫描(CT)异常,此外,遗传性PVOD患者的特点是发病年龄比没有EIF2AK4双列变异的PVOD患者早。
PAH的基因测试的现状
基因检测可以帮助临床医生更好地描述PAH患者的表型,识别可能被错误分类的患者,促进适当的管理。考虑到具有BMPR2致病变体的个体,平均来说,在较小的年龄就患上了PAH,呈现出更多的血流动力学特征,具有更高的死亡或肺移植风险,基因诊断可能对临床管理和治疗策略产生重大影响。例如,可能会建议更频繁的监测,以允许早期治疗升级和/或开始使用联合治疗。此外,基因检测有助于对家庭成员进行风险分层,它可能允许进行生殖选择,如植入前基因检测。
PVOD/PCH可能难以诊断,其特点是放射学异常、低DLCO和对PAH治疗的反应差。PVOD/PCH可能被诊断不足,因为在疾病的早期阶段,明显的特征可能并不明显。双链EIF2AK4致病变体至少占所有PVOD/PCH病例的25%。基因检测可以在被误诊为IPAH的患者中识别出双拷贝EIF2AK4变体。由于PVOD/PCH患者的预后很差,并且在PAH治疗中会出现肺水肿,基因检测可以识别这些被错误分类的患者,从而进行适当的管理和早期转诊为肺移植,因为有报道称EIF2AK4患者的疾病进展很快。PVOD/PCH在近亲家庭中也被描述得更为频繁。
遗传性出血性毛细血管扩张症(HHT)(或OslerWeber-Rendu病)患者PAH的致病基因:ACVRL1,ENG和SMAD4,这些基因也属于BMP-TGF-β家族。HHT的主要特征是 粘膜毛细血管扩张,复发性鼻衄,大面积的动静脉畸形和。在罕见的情况下,还包括多发性硬化症。HHT是常染色体显性遗传,在青春期前后发病。到60岁时,HHT的外显率基本上是完全的,但PAH的外显率要低得多。只有极少数HHT易感变异体携带者在60岁以上有PAH,但没有任何HHT的迹象。相反,与ACVRL1突变相关的PAH的特点是发病年龄中位数为20岁,因此,PAH可能是这些年轻患者随后发展为HHT的第一个明显迹象。事实上,基因检测可以识别这些患者,并促进患者及其亲属对HHT并发症(动静脉畸形)的识别。
一些易感的PAH基因也与儿童和成人的肺部发育异常有关,如T-box蛋白4(TBX4)。该基因以前与伴有或不伴有PAH的小髌骨综合征有关,是常染色体显性遗传,具有不完全外显率和可变的表现力。在小儿PAH中已经观察到致病性TBX4变体的富集。基因诊断可以帮助评估相关的髋关节和膝关节问题。同样,在PAH之外的间质性肺病患者中,也发现了编码血管内皮生长因子受体2的基因KDR的蛋白截断变体。在这些情况下,PAH可能与低DLCO(KDR)或支气管异常(TBX4)有关。基因检测可能有助于将这些患者与慢性肺部疾病引起的第3组PH区分开来,特别是如果低DLCO与过去的吸烟史一起出现。
在患有先天性心脏病的APAH患者中,特别是在患有这种疾病的儿童中,已发现3%-7%的患者存在转录因子SOX17的致病变异。这些患者大多表现为简单的心脏缺陷,如动脉间隔缺陷、动脉导管未闭、卵圆孔未闭和室间隔缺陷。此外,在一部分患者中还出现了胸部CT的异常,如肺血管扩张、迂回、磨玻璃状不透明和咯血。虽然致病的SOX17变体在先天性心脏病APAH中更为常见,但在家族性和IPAH中也有描述。同一家族的PAH患者中甚至发现了相同的SOX17变异体,这些患者有的伴有先天性心脏病,有的没有。同样,CHD-APAH患者的致病变体不仅在SOX17中被发现,也在其他PAH基因中被发现,如TBX4和BMPR2。
对于药物和毒素诱发的PAH患者,几乎没有证据。目前的证据表明,只有曾经摄入厌食原的患者可能存在致病变体,因此应进行遗传咨询和检测。对于其他特定的药物或毒素暴露,只有少数病例有致病变体的报道。在大多数其他病人中,除了药物或毒素暴露外,没有发现易感的遗传变异。例如,在20多名摄入Colza油并随后发展为多发性硬化症的患者中,无法确定致病变体。同样,在13名PVOD患者的子集中,以前有中度或高度的三氯乙烯暴露,但无法确定致病变体,而在12名PVOD患者中,有5名没有暴露于同一物质或只有轻微的暴露,是双亲EIF2AK4变体的携带者。
我们的建议是,至少应对有PAH家族史的患者、IPAH患者、厌食症诱发的PAH患者和先天性心脏病APAH患者进行基因检测。先天性心脏病APAH患者。对疑似或确诊的PVOD/PCH和其他发育性肺部疾病的患者也应提供遗传咨询和检测。根据美国医学遗传学和基因组学学院和分子病理学协会(ACMG)的指南,应向具有已确定的致病/可能致病变体的PAH索引病例的亲属提供级联基因测试。目前还没有足够的证据推荐对第2-5组的肺动脉高压患者进行基因检测。只有当肺动脉高压有家族聚集性或任何考虑上述鉴别诊断时,才应提供遗传学。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言