


Viruses 2024, 16(5), 658; https://doi.org/10.3390/v16050658 - 23 Apr 2024
Abstract
Recombinant adenoviruses are widely used in clinical and laboratory applications. Despite the wide variety of available sero- and genotypes, only a fraction is utilized in vivo. As adenoviruses are a large group of viruses, displaying many different tropisms, immune epitopes, and replication characteristics,
[...] Read more.
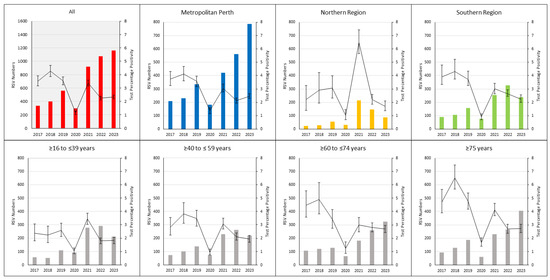
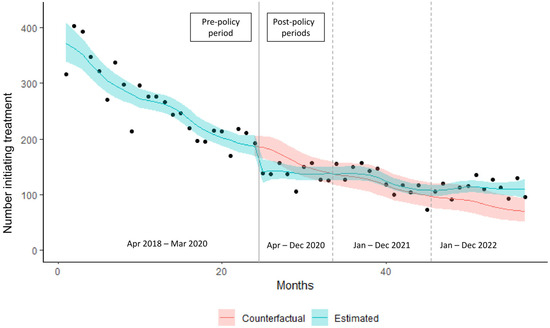
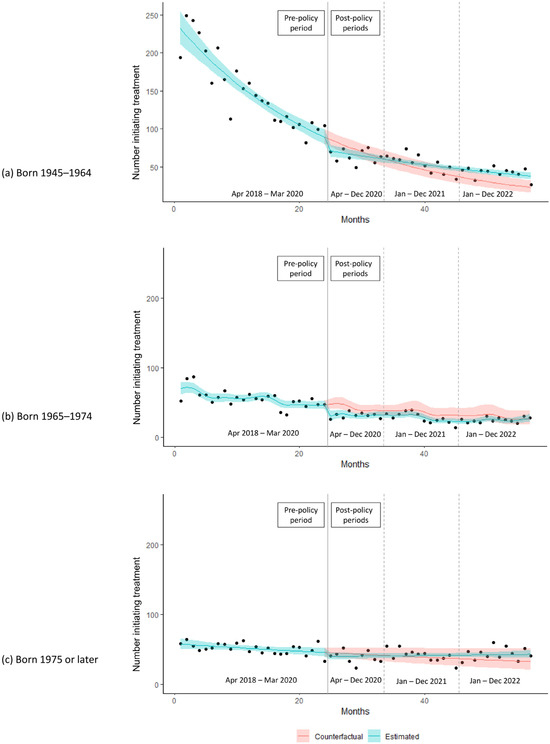
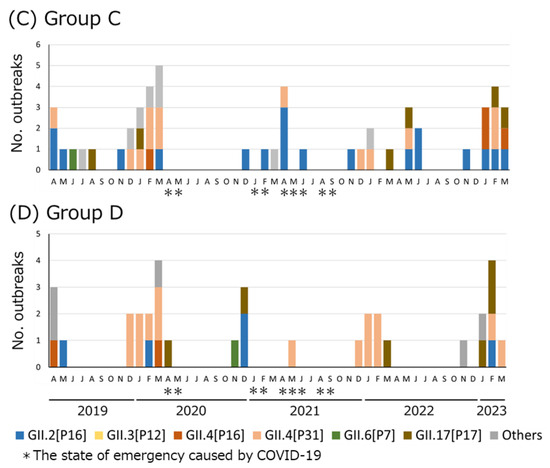
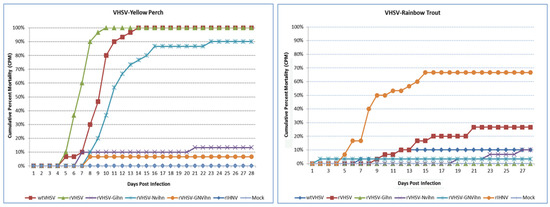
Recombinant adenoviruses are widely used in clinical and laboratory applications. Despite the wide variety of available sero- and genotypes, only a fraction is utilized in vivo. As adenoviruses are a large group of viruses, displaying many different tropisms, immune epitopes, and replication characteristics, the merits of translating these natural benefits into vector applications are apparent. This translation, however, proves difficult, since while research has investigated the application of these viruses, there are no universally applicable rules in vector design for non-classical adenovirus types. In this paper, we describe a generalized workflow that allows vectorization, rescue, and cloning of all adenoviral species to enable the rapid development of new vector variants. We show this using human and simian adenoviruses, further modifying a selection of them to investigate their gene transfer potential and build potential vector candidates for future applications.
Full article
(This article belongs to the Special Issue 15th International Adenovirus Meeting)
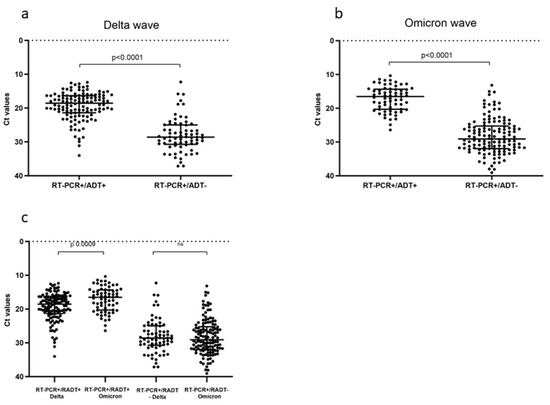
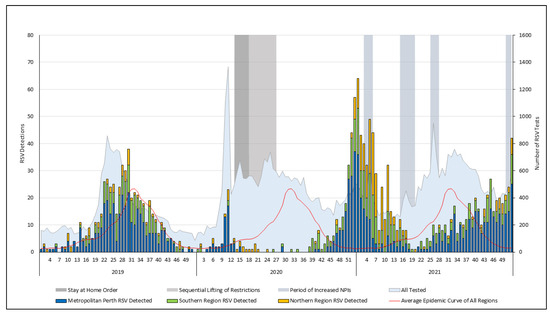
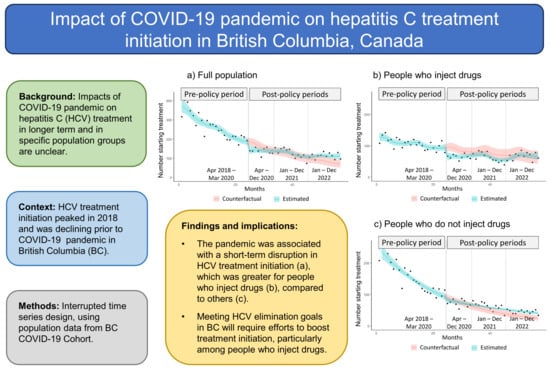

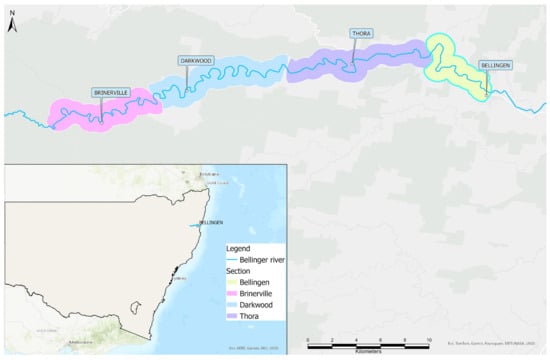

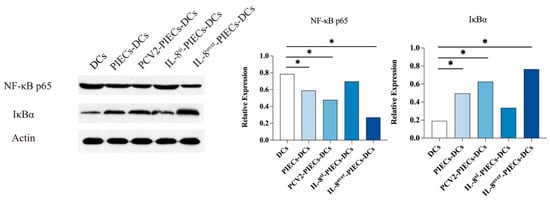
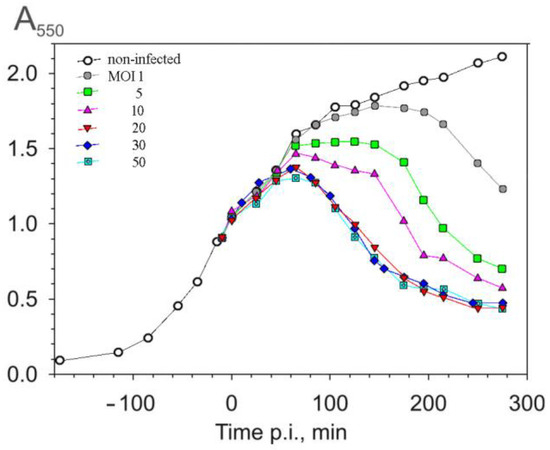
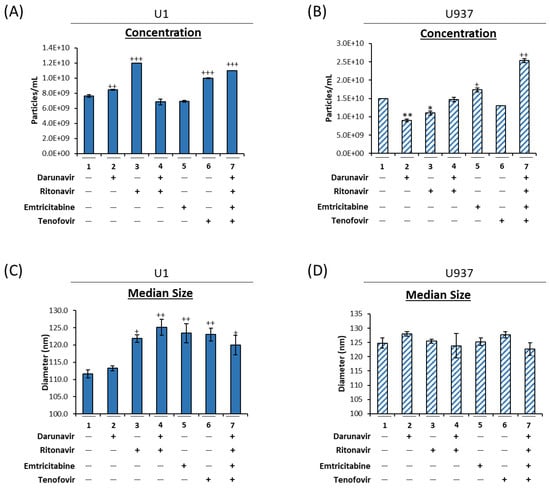


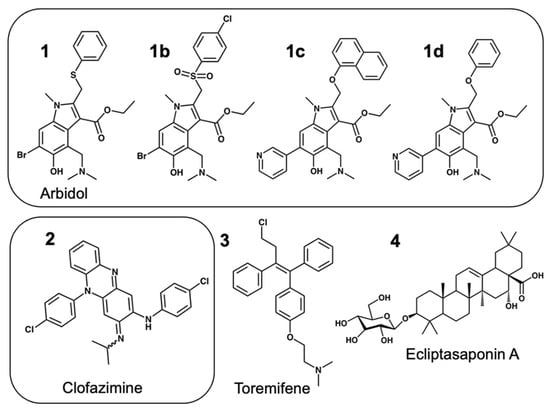





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}