


Genes 2024, 15(5), 566; https://doi.org/10.3390/genes15050566 (registering DOI) - 27 Apr 2024
Abstract
Soybean mosaic virus (SMV) is one of the main pathogens that can negatively affect soybean production and quality. To study the gene regulatory network of soybeans in response to SMV SC15, the resistant line X149 and susceptible line X97 were subjected to transcriptome
[...] Read more.
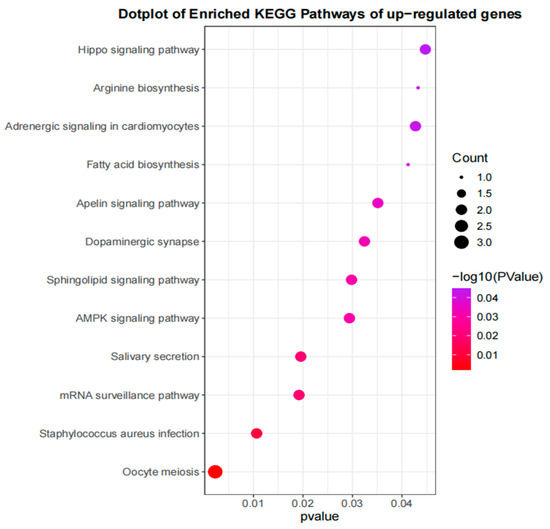
Soybean mosaic virus (SMV) is one of the main pathogens that can negatively affect soybean production and quality. To study the gene regulatory network of soybeans in response to SMV SC15, the resistant line X149 and susceptible line X97 were subjected to transcriptome analysis at 0, 2, 8, 12, 24, and 48 h post-inoculation (hpi). Differential expression analysis revealed that 10,190 differentially expressed genes (DEGs) responded to SC15 infection. Weighted gene co-expression network analysis (WGCNA) was performed to identify highly related resistance gene modules; in total, eight modules, including 2256 DEGs, were identified. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of 2256 DEGs revealed that the genes significantly clustered into resistance-related pathways, such as the plant–pathogen interaction pathway, mitogen-activated protein kinases (MAPK) signaling pathway, and plant hormone signal transduction pathway. Among these pathways, we found that the flg22, Ca2+, hydrogen peroxide (H2O2), and abscisic acid (ABA) regulatory pathways were fully covered by 36 DEGs. Among the 36 DEGs, the gene Glyma.01G225100 (protein phosphatase 2C, PP2C) in the ABA regulatory pathway, the gene Glyma.16G031900 (WRKY transcription factor 22, WRKY22) in Ca2+ and H2O2 regulatory pathways, and the gene Glyma.04G175300 (calcium-dependent protein kinase, CDPK) in Ca2+ regulatory pathways were highly connected hub genes. These results indicate that the resistance of X149 to SC15 may depend on the positive regulation of flg22, Ca2+, H2O2, and ABA regulatory pathways. Our study further showed that superoxide dismutase (SOD) activity, H2O2 content, and catalase (CAT) and peroxidase (POD) activities were significantly up-regulated in the resistant line X149 compared with those in 0 hpi. This finding indicates that the H2O2 regulatory pathway might be dependent on flg22- and Ca2+-pathway-induced ROS generation. In addition, two hub genes, Glyma.07G190100 (encoding F-box protein) and Glyma.12G185400 (encoding calmodulin-like proteins, CMLs), were also identified and they could positively regulate X149 resistance. This study provides pathways for further investigation of SMV resistance mechanisms in soybean.
Full article
(This article belongs to the Section Plant Genetics and Genomics)








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}