
【协和医学杂志】首发症状为皮肤肿物、皮疹、骨痛的原发性肝脏神经内分泌肿瘤一例
2023-08-14 协和医学杂志 协和医学杂志 发表于上海

本例患者高龄,病情复杂难以确诊,且进展迅速、累及多系统,明确诊断时已无法耐受手术治疗,从初诊至死亡仅2个月,唯有初期快速识别疾病,才有可能赢得治疗时机。
患者女性,71岁,因“左足肿物、皮疹、骨痛1个月”于2022年3月4日收住青岛大学附属医院创伤外科。
2022年2月,患者左足跟出现紫红色、类圆形肿物,直径约6 cm,高于皮面(图1A),伴双侧小腿紫红色丘疹(图1B),左下肢骨痛,以膝关节、足背为主,活动受限。后肿物破溃、渗出,分泌物呈黄色脓样,为进一步诊治就诊于青岛大学附属医院创伤外科。患者既往体健,个人史、月经史、婚育史、家族史无特殊。
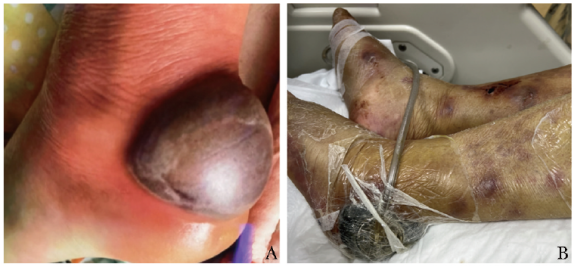
图1A 患者左足紫红色半球形肿块;图1B 双下肢紫红色丘疹
入院查体:体温36.3 ℃,心率94次/min,呼吸频率20次/min,血压140/80 mm Hg(1 mm Hg=0.133 kPa),神志清,心肺未见异常、腹部平软。左足外踝红肿、破溃渗出,伴右侧足背及双侧小腿紫红色丘疹,左下肢骨压痛,以膝关节及足背为著。
辅助检查:白细胞21.09×109/L,中性粒细胞比率0.88,C反应蛋白 174.56 mg/L,降钙素原 0.43 μg/L。左踝关节MR平扫示左足跟骨异常信号,左踝关节积液伴周围软组织肿胀;右足MRI示右足距骨、跟骨、第3、5跖骨异常信号,右踝关节积液伴周围软组织肿胀(图1C)。
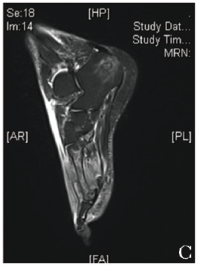
图1C 患者MRI示右足跟骨、第3、5跖骨病变,周围软组织肿胀
患者左足跟肿物伴破溃渗出1个月,分泌物呈黄色脓样,伴同侧肢体骨痛,影像学提示软组织肿胀、骨异常信号,炎症指标升高,入院后出现发热,初步考虑软组织感染、感染性骨髓炎可能性大,拟行清创术,术前完善一般辅助检查。
入院后给予哌拉西林钠/他唑巴坦钠抗感染治疗。
辅助检查:白蛋白27.9 g/L,空腹血糖 11.92 mmol/L。常见女性肿瘤标志物筛查(包括特异β人绒毛膜促性腺激素,鳞状细胞癌相关抗原,癌胚抗原,甲胎蛋白,神经元特异性烯醇化酶,糖类抗原72-4、19-9、125、15-3,胃泌素释放肽前体)未见明显异常。
胸部CT示双肺炎症可能性大,肝内低密度、部分肝内胆管扩张。上腹CT增强扫描、肝MRI增强扫描均提示肝内占位性病变,大小约为123 mm×94 mm,动脉期明显强化,静脉、延迟期逐渐减弱,可见强化假性包膜。
术前检查提示肝恶性肿瘤可能性大,结合患者病史,既往体健,发病后出现发热、皮疹、骨痛、血糖升高、低蛋白血症,考虑不排除神经内分泌肿瘤(NET),采用18F-AlF-NOTA-octreotide(18F-OCT)与18F-FDG PET/CT联合显像检测NET准确性较高[1]。由于患者出现发热、炎症指标升高、血糖升高、营养状态差、合并肺部感染等症状、一般状况差,决定优先给予抗感染治疗及清创手术,同时行PET/CT检查,待病情稳定后再行肝穿刺活检。
给予哌拉西林钠/他唑巴坦钠联合利奈唑胺治疗,炎症指标持续升高,再次调整治疗方案,给予亚胺培南西司他丁钠、利奈唑胺及万古霉素联合治疗,尿培养示假丝酵母菌,加用氟康唑抗感染治疗。
患者存在肺部感染、胸腔积液,行右侧胸腔积液穿刺引流术;全身皮下触及多发结节(图1D),质硬,行穿刺培养;考虑为“感染性骨髓炎”,行骨髓穿刺常规检查、免疫组化检查及标本培养,检查结果未见明显异常;行左足感染病灶清除术+真空辅助闭合负压引流术,清除坏死皮肤及皮下组织,术中可见跟骨外侧坏死骨皮质碎屑,刮除坏死骨皮质,可见骨松质内大量白色颗粒,一并清除后,彻底冲洗。术后转入ICU。

图1D 患者PET/CT示全身广泛皮下结节
辅助检查:血液、左足脓肿引流液、骨髓、皮下结节穿刺物等标本培养及各标本病原微生物二代测序(NGS)结果均为阴性。足跟软组织病灶活检病理示皮肤组织慢性炎症伴溃疡形成及肉芽组织增生(图1E),表皮假上皮瘤样增生,真皮及皮下组织急慢性炎症细胞及泡沫细胞浸润并多核巨细胞反应(图1F),脂肪细胞坏死,小血管硬化,考虑为“炎性病变”。
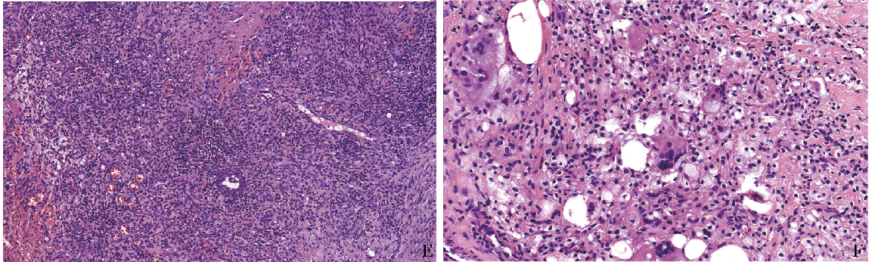
图1EF 患者皮肤组织急慢性炎伴溃疡形成及肉芽组织增生(HE染色,×200);皮下组织急慢性炎性细胞及泡沫细胞浸润并多核巨细胞反应
特殊染色:抗酸染色(AFB)(-),六胺银染色(GMS)(+),黏液卡红染色(-),革兰氏染色(GS)(-),过典酸雪夫氏染色(PAS)(-)。分子检测示PCR-TB阴性。跟骨病灶:粉染无结构坏死物及死骨组织。
18F-OCT示肝左叶低密度肿块,边缘清晰,内多发条状走行低密度影,略呈辐辏样分布,肿块未见明显示踪剂摄取增高,未见生长抑素受体表达;右侧顶部皮下、双侧背部皮下、右侧胸壁皮下、双侧臀部及双下肢皮下多发大小不等软组织结节,部分见示踪剂轻度摄取增高,最大标准摄取值(SUVmax)约为1.7(图2A)。
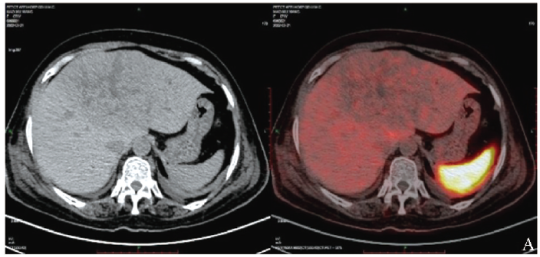
图2A 患者18F-OCT示肝左叶低密度肿块,未见明显示踪剂摄取增高
18F-FDG PET/CT示肝左叶巨大低密度肿块,代谢异常增高,SUVmax约为9.1;中轴骨及双侧近段四肢骨弥漫性轻度代谢增高(图2B)。结合免疫组化及18F-OCT结果,符合NET G3诊断。
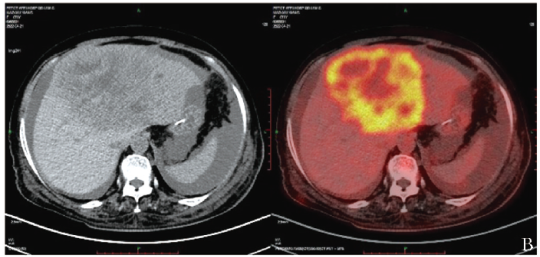
图2B 患者18F-FDG PET/CT示肿块代谢异常增高,SUVmax约为9.1
NET分泌功能检测:胰岛素74.61 pmol/L(正常范围:17.8~173 pmol/L),C-肽 2.25 nmol/L(正常范围:0.37~1.47 nmol/L),胰高血糖素 9.35 pmol/L(正常范围:1.5~18 pmol/L),人嗜铬粒蛋白A 796.72 μg/L(正常范围:27~94 μg/L),5-羟吲哚乙酸 3.0 mg/g Cr(≤0 mg/g Cr),高香草酸1.8 mg/g Cr(<8 mg/g Cr),香草扁桃酸 2.9 mg/g Cr(<7 mg/g Cr),肌酐0.46 g/L(正常范围:0.2~3.2 g/L)。
患者血液、引流液、骨髓、皮下结节穿刺等标本培养及NGS均未发现致病菌,且抗生素治疗效果差,病理提示炎性细胞浸润,考虑皮肤肿物、皮下结节及骨髓炎为无菌性炎症,根据18F-OCT与18F-FDG PET/CT联合显像考虑肝NET可能性大,综上,拟诊为NET合并副肿瘤综合征。为进一步明确诊治,行肝穿刺活检。
肝穿刺病理示恶性肿瘤(图3A),免疫组化染色示突触素(Syn)(+)(图3B)、 嗜铬粒蛋白A(CgA)(+)(图3C)、 生长抑素受体2(SSTR2)(-)、CDX-2(-)、 Ki-67指数(+,约40%)、p53(+,约5%,野生型)、 Rb(-)、Hepatocyte(-)、GPC3(-)、Arginase-1(-)、GS(-)、HSP70(-)、CD10(-)、CK7(+)、AFP(-)。
综上,诊断为NET G3。
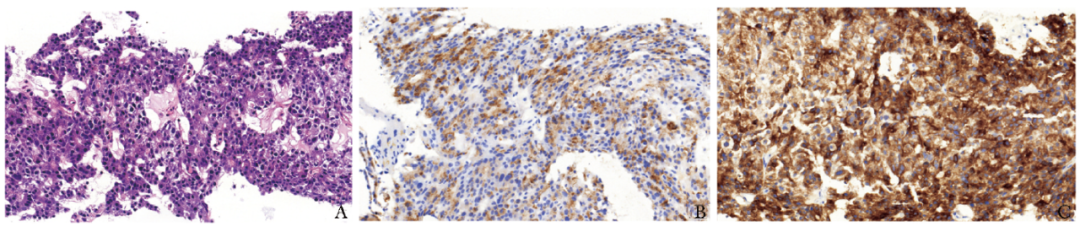
图3 患者肝脏病理及免疫组化染色
A.肿瘤细胞大小较一致,核深染,胞浆呈颗粒状,核分裂像易见(HE染色,×200);B.肿瘤细胞突触素(+)(EnVision染色,×200);C.肿瘤细胞嗜铬粒蛋白A(+)(EnVision染色,×400)
根据PET/CT联合显像及病理与免疫组化结果,符合NET G3诊断。NET多见于胃肠道、胰腺等器官,肝脏是最常见的转移性器官,而原发于肝脏的神经内分泌肿瘤(PHNET)十分罕见,目前病理和免疫组化染色无法区分原发性和转移性肝脏NET,均建议排除转移瘤后诊断为PHNET,本例患者行全面影像学检查后未发现其他可能原发灶,考虑诊断为PHNET,且伴有皮肤肿物、皮疹、无菌性骨髓炎等副肿瘤综合征表现。手术切除是早期PHNET最有效的治疗方式,部分患者有望完全治愈,获得良好的长期无瘤生存,也可缓解部分患者的副肿瘤综合征表现。
患者间断高热,炎症指标高,痰培养示鲍氏不动杆菌、肺炎克雷伯菌阳性,继续给予抗感染治疗;血气分析示呼吸衰竭,给予气管插管、呼吸机辅助通气;少尿,间断行连续性肾脏替代治疗(CRRT);辅以营养支持、输注白蛋白、控制血糖、控制心室率、化痰等对症支持治疗。拟行肝脏病损切除术,全身麻醉后患者出现血压、血氧饱和度急剧下降,给予扩容、升压、纠酸等抢救措施后生命体征恢复平稳,手术暂停返回ICU。
患者病情逐渐进展,间断嗜睡状态,炎症指标持续升高,皮下结节发展至全身,部分高出皮面形成紫红色包块,表面破溃伴脓样渗出,合并多器官功能障碍(肝脏、呼吸、肾脏、血液、循环)逐渐加重。2022年5月3日患者进入昏睡状态,血压难以维持,抢救无效死亡。本例患者最终诊断包括:PHNET、副肿瘤综合征、无菌性骨髓炎。
本例患者以皮肤肿物、皮疹、骨痛起病,继而出现血糖升高、低蛋白血症,检查后发现肝脏占位性病变,经18F-OCT与18F-FDG PET/CT联合显像提示NET G3,并最终根据病理与免疫组化结果确诊。
NET是一种起源于全身弥漫分布的神经内分泌细胞异质性肿瘤,发病率约为6.98/100 000 [2],其中PHNET约占NET的0.3%,较为罕见,多为中老年发病,男女比例相当[3],早期多无症状,中晚期或肿瘤体积较大时出现腹痛、腹部包块、体质量减轻等症状,神经内分泌症状较少见[4],需根据病理活检及免疫组化(标志物包括CgA、Syn、神经元特异性烯醇化酶、CD56)结果进行诊断[5]。如活检难以获取,可行影像学检查以辅助诊断,但CT、MRI等传统影像学技术因缺乏特异性,诊断NET较困难。
有研究表明,18F-OCT与18F-FDG联合显像可提高NET及转移灶的诊断准确性,大部分NET细胞表面表达SSTR,18F-OCT可用于体内SSTR高表达恶性肿瘤的特异性检测:大部分高分化NET细胞生长缓慢,葡萄糖代谢率低,但表面过表达SSTR 2,可高特异性结合18F-OCT;低分化NET细胞表面已失去原有特征,SSTR表达量下降,但侵袭性更高,对18F-FDG摄取能力更强,因此,18F-OCT与18F-FDG PET/CT联合显像能更准确地检测NET,本研究进一步验证了其诊断价值[1]。由于肝脏被认为是 NET最常见的转移部位,因此 PHNET 的诊断应排除转移性NET。
本例患者首发症状不典型,发热、皮损红肿热痛伴脓样渗出、骨痛、炎症指标升高,极易被误诊为感染性病变,反复病原学检测阴性,病理示炎性细胞浸润,考虑为无菌性炎症,结合PHNET最终诊断为副肿瘤综合征,无菌性骨髓炎属除外性诊断,需重点与感染、肿瘤进行区分,本例患者是在病原学及组织病理学检查完善后明确诊断,但仍需讨论存在特殊感染的可能。
本例患者表现为肝占位合并皮肤损害,需与以下疾病相鉴别:
(1)默克尔细胞癌(MCC)伴肝转移,MCC是一种罕见的皮肤NET,表现为单发无痛性紫红色结节,多发生于头颈部,其次为四肢及臀部,根据病理及免疫组化进行诊断,典型病理特征是密集排列的基底样细胞形成的层状或巢状皮肤肿瘤,神经丝蛋白、CK-20、CK-7和甲状腺转录因子-1具有很高的灵敏度和特异性[6]。本例患者通过皮肤病理可排除该诊断。
(2)脂肪酶过度分泌综合征(LHS)。LHS是胰腺癌特有的副肿瘤综合征,主要特征为皮下痛性结节,多发生于小腿,表现为肉色至紫褐色,骨与关节均可受累。LHS多发生于伴有肝转移的胰腺癌中,胰腺肿块通常大、圆、界限分明[7],本例患者无胰腺病变,可排除该诊断。
治疗方面,手术切除是 PHNET 的主要治疗方式。Li等[3]研究表明,76例接受手术治疗的患者5年生存率为71.9%,疗效确切,但多项研究表明,术后复发率高达20~40%[8-10],因此术后需建立密切随访。其他治疗方式包括肝动脉化疗栓塞术、射频消融、经皮乙醇注射治疗、肝移植、奥曲肽等生长抑素类似物和化疗,但其作用及有效性尚不清楚,需要进一步研究。
本例患者高龄,病情复杂难以确诊,且进展迅速、累及多系统,明确诊断时已无法耐受手术治疗,从初诊至死亡仅2个月,唯有初期快速识别疾病,才有可能赢得治疗时机。
综上,对于老年患者出现不明原因的突发难治性皮肤肿物、皮疹、骨痛,需警惕副肿瘤综合征;如患者出现骨痛,组织病原学检查结果为阴性,影像学提示骨异常信号、代谢增高,活检见骨坏死,未见肿瘤性病变,则符合无菌性骨髓炎表现;如患者肝占位伴有发热、皮肤肿物、皮疹、骨痛、血糖升高、低白蛋白等表现,需考虑NET,并进一步明确是否肝脏原发,18F-OCT与18F-FDG PET/CT联合显像具有重要价值,有助于明确NET诊断。
参考文献:
[1]陈博, 冯洪波, 宫晓艳, 等. 18F-AlF-NOTA-octreotide联合18F-FDG PET/CT显像用于神经内分泌瘤[J]. 中国医学影像技术, 2021, 37: 663-668.
[2]Cives M, Strosberg JR. Gastroenteropancreatic Neuroendocr-ine Tumors[J]. CA Cancer J Clin, 2018, 68: 471-487.
[3]Li YF, Zhang QQ, Wang WL. Clinicopathological Characteristics and Survival Outcomes of Primary Hepatic Neuroendocrine Tumor: A Surveillance, Epidemiology, and End Results (SEER) Population-Based Study[J]. Med Sci Monit, 2020, 26: e923375.
[4]陈超, 牛忠锋, 施伟, 等. Ⅰ型神经纤维瘤病合并肝脏原发神经内分泌肿瘤一例[J]. 中华医学杂志, 2021, 101: 1535-1536.
[5]刘哲, 李晓明, 蔡守旺, 等. 原发性肝脏神经内分泌肿瘤误诊分析[J]. 中华转移性肿瘤杂志, 2020, 3: 52-56.
[6]Becker JC, Stang A, DeCaprio JA, et al. Merkel cell carcinoma[J]. Nat Rev Dis Primers, 2017, 3: 17077.
[7]Taskin OC, Adsay V. Lipase hypersecretion syndrome: A distinct form of paraneoplastic syndrome specific to pancreatic acinar carcinomas[J]. Semin Diagn Pathol, 2019, 36: 240-245.
[8]Shi C, Zhao Q, Dai B, et al. Primary hepatic neuroend-ocrine neoplasm: Long-time surgical outcome and prognosis[J]. Medicine (Baltimore), 2018, 97: e11764.
[9]Iwao M, Nakamuta M, Enjoji M, et al. Primary hepatic carcinoid tumor: case report and review of 53 cases[J]. Med Sci Monit, 2001, 7: 746-750.
[10]Knox CD, Anderson CD, Lamps LW, et al. Long-term survival after rep for primary hepatic carcinoid tumor[J]. Ann Surg Oncol, 2003, 10: 1171-1175.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



