儿童颅内非典型畸胎样/横纹肌样瘤2例
2018-10-12 苏建云 张刚 杨张凯 中国临床神经外科杂志

非典型畸胎样/横纹肌样瘤(atypical teratoid/rhabdoid tumor,AT/RT)属于胚胎性肿瘤之一,由Biggs等首次报道,发病率极低,占儿童颅内肿瘤1%~3%,70%~90%以上发病于低龄儿童(<3岁)。AT/RT 恶性度极高,预后极差,中位生存期0.5~1年。2014年收治2例儿童颅内AT/RT,现报道如下。
非典型畸胎样/横纹肌样瘤(atypical teratoid/rhabdoid tumor,AT/RT)属于胚胎性肿瘤之一,由Biggs等首次报道,发病率极低,占儿童颅内肿瘤1%~3%,70%~90%以上发病于低龄儿童(<3岁)。AT/RT 恶性度极高,预后极差,中位生存期0.5~1年。2014年收治2例儿童颅内AT/RT,现报道如下。
1. 病例资料
病例1:男,1岁2个月,因精神差伴间断性头部后仰强直10 h急诊入院。入院时体格检查:颈部活动稍强直,余未见明显阳性体征。头颅CT示后颅窝占位(图1A),位于第四脑室内,邻近脑干背侧,呈偏心囊性及边界实质性改变,大小约4 cm×5 cm×4 cm,伴瘤周出血、水肿以及不规则斑点状钙化影。头颅MRI示肿瘤呈长T1、长T2信号,边界有不规则强化影(图1B~D)。
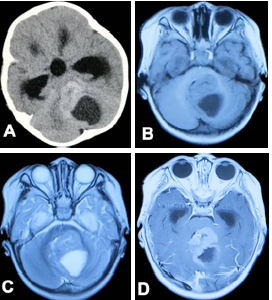
图1 颅后窝非典型畸胎样/横纹肌样瘤影像学表现A. CT 示颅后窝囊实性占位,偏心囊性变,周边实质组织出血伴钙化;B、C. MRI 示颅后窝小脑半球间长T1、长T1囊性区,稍高T1、等T2信号;D. MRI 增强示实质区不规则花边样强化,边界尚清晰
血清肿瘤标志物甲胎蛋白(α-fetoprotein,AFP)、癌胚抗原(carcino-embryonicantigen,CEA)以及人绒毛膜促性腺激素β(human chorionic gonadotropin beta,β-HCG)均正常,全组染色体核型分析示染色体形态大致正常。采取标准后颅窝占位性病变切除术治疗。术中见肿瘤实质呈灰红色、质软、血供丰富,囊腔内见淡黄血性囊液,肿瘤边界尚清晰,术中肿瘤达肉眼完整切除。术后病理结果明确为AT/RT(WHO Ⅵ级)。术后CT复查示肿瘤完整切除。术后6个月影像学复查见局部肿瘤复发,但患儿未见明显临床症状,家属拒绝行进一步放疗。
病例2:男,4岁9个月,因间断呕吐3 d、癫痫发作6 h急诊入院。入院时体格检查神经系统查体未见明显阳性体征。头颅CT示右侧颞叶以及侧脑室下角内混杂高密度影,内可见不规则点状钙化(图2A)。头部MRI示右侧颞叶、小脑半球、额颞叶皮层多灶性稍长T1、长T2 信号影,呈非均质性强化(图2B~D)。
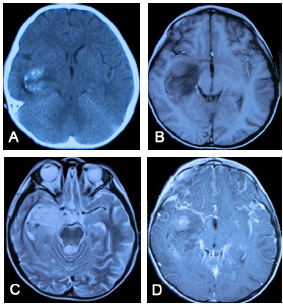
图2 颅内多发非典型畸胎样/横纹肌样瘤影像学表现E. CT 示右侧颞叶、侧脑室下角混杂高密度,内可见散在点状钙化影;F、G. MRI 示肿瘤呈稍长T1、长T2信号;H. MRI增强示团块状非均质性强化
血清肿瘤标志物AFP、CEA以及β-HCG均正常。采用左侧颞顶入路手术。术中见颞极皮层灰白色改变,瘤体广泛皮层种植转移,切开颞中回皮层后见瘤体呈豆腐渣、灰白色胶冻样,边界欠清晰,大小约3 cm×5 cm×3 cm,术中大部分切除瘤体。术后病理结果明确为AT/RT(WHO Ⅵ级)。术后3 d家属选择院外治疗,因癫痫持续状态而死亡。
2. 讨论
AT/RT颅后窝多见,约占半数以上,此外桥小脑脚、大脑皮层、松果体以及脊髓亦见生长;90%以上Ini-1 表达阴性。本文病例1肿瘤位于颅后窝,为AT/RT好发部位,影像学检查显示瘤体呈囊实性改变。术前影像资料提示髓母细胞瘤或原始神经外胚层肿瘤可能。髓母细胞瘤为小儿颅后窝占半数以上恶性肿瘤,幕上罕见,而AT/RT 钙化、出血更为常见。原始神经外胚层肿瘤影像学囊性变及坏死、钙化可分布于整个肿瘤,这与AT/RT较大偏心性囊性变不同。AT/RT光镜可见典型的细胞核,深染色的细胞质,有丝分裂及细胞坏死多见,细胞簇状分布,形态较大,呈梭形或多角形;电镜下显示细胞质内可见“螺旋状”神经纤丝成分,提示内含有波形蛋白(vimentin,Vim)。AT/RT组织上皮膜抗原(epithelial membrane antigen,EMA)、Vim及部分平滑肌肌动蛋白表达阳性,而髓母细胞瘤或原始神经外胚层肿瘤常表达阴性。
本文病例2肿瘤位于幕上,呈实质性改变,病灶位于侧脑室后角以及下角,就诊时已发生颅内多发转移。文献报道超过20%的AT/RT有肿瘤细胞循脑脊液播散,因此部分病人脑脊液可以检测到典型的富含特异细胞核呈簇状分布的巨型细胞以及微小型肿瘤细胞,对于诊断AT/RT具有重要意义。病例2肿瘤位于幕上室管膜瘤好发部位,CT平扫可见等或者稍高密度影,MRI平扫可见稍长T1、长T2信号,增强扫描呈均匀强化高信号影,与室管膜瘤极其相似。室管膜瘤光镜下可见细胞呈片状分布,局部可见血管周围假菊团征象,细胞呈假复层腺样或者裂隙样排列,免疫组化结果提示菊形团细胞胶质纤维酸性蛋白(+)、S-100以及Vim(+),部分瘤细胞EMA(+),且突触素以及神经元特异性烯醇化酶表达阴性。
Nobusawa等指出AT/RT与室管膜瘤可能有着相同的组织起源与致病机制,融合基因C11orf95-RELA作为室管膜瘤重要致病基因标志,亦可在AT/RT肿瘤中表达。胶质瘤呈不规则环形强化,肿瘤中心囊性变常见,但瘤周水肿更为明显,这与AT/RT轻度的瘤周水肿不同。此外,胶质瘤有明显的纤丝蛋白成分且散在分布,星形细胞胶质瘤有双核以及透明样细胞质成分特点,这与AR/RT在光镜下细胞形态有所差别,且例2肿瘤标记S-100以及Olig-2表达阴性。
神经元特异性核蛋白在正常皮层神经元中表达,AT/RT中该抗原表达阴性,与Olig-2阴性表达结合,亦基本排除少突胶质细胞瘤。海绵状血管瘤CT平扫示圆形或者小片状高密度影,病灶中心因钙化呈斑点状钙化影,瘤周因反复出血致含铁血黄素沉积呈短T1、长T2信号“黑环征”,进一步SWI检查示低信号改变,可辅助诊断。儿童脑肿瘤中,AT/RT为首个有明确致病基因位点的肿瘤。现已明确90%以上AT/RT 病人22q(q13;q11.2;q14.2)染色体基因突变或者缺失,从而导致Ini-1蛋白表达阴性,是肿瘤发生的关键病理机制。而少数病例Ini-1亦可表达阳性,其可能通过转录后调控因子作用来起致瘤作用。
本文病例1肿瘤Ini-1阴性,与少见的环状22号染色体形态缺失不同,需进一步行染色体荧光杂交技术进行突变基因位点确认。即使术中完整切除肿瘤联合后期放、化疗,AT/RT预后仍不理想,平均生存期为3年。本文病例1术中完整切除肿瘤,对于解除梗阻性脑积水意义重大,尽管术后6个月肿瘤复发,但未见明显临床症状。病例2就诊时已发生颅内多灶转移,术中见肿瘤分界不清,大部分切除,因此进一步术后积极放、化疗显得极为重要。
Lafay等报道高剂量化疗(甲氨蝶呤、塞哌嗪)联合自体干细胞移植较传统化疗而言可延长患儿生存时间,但其有效性以及安全性仍需进一步证实。顺铂作为AT/RT术后化疗的主要药物之一,但化疗抵抗以及肿瘤复发较为常见,可能机制与通过调控STAT3/Snail信号通路,使肿瘤产生新的免疫表型,最终导致治疗失败。儿童颅内AT/RT肿瘤单纯手术治疗可以延缓疾病进展,后期应积极选择有效放、化疗以期改善患儿预后。
原始出处:
苏建云,张刚,杨张凯,李楠,王蕊,陈广生,史航宇.儿童颅内非典型畸胎样/横纹肌样瘤2例并文献复习[J].中国临床神经外科杂志,2017,22(11):777-779.
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#非典型#
43