靶向p53突变的药物研发进展
2023-03-25 精准药物 网络 发表于上海

目前RAS已经有两款药物上市,打破它的“不可成药性”观点,相信不久的将来mutp53也一定会成为可成药的靶点。

wtp53和mutp53的功能
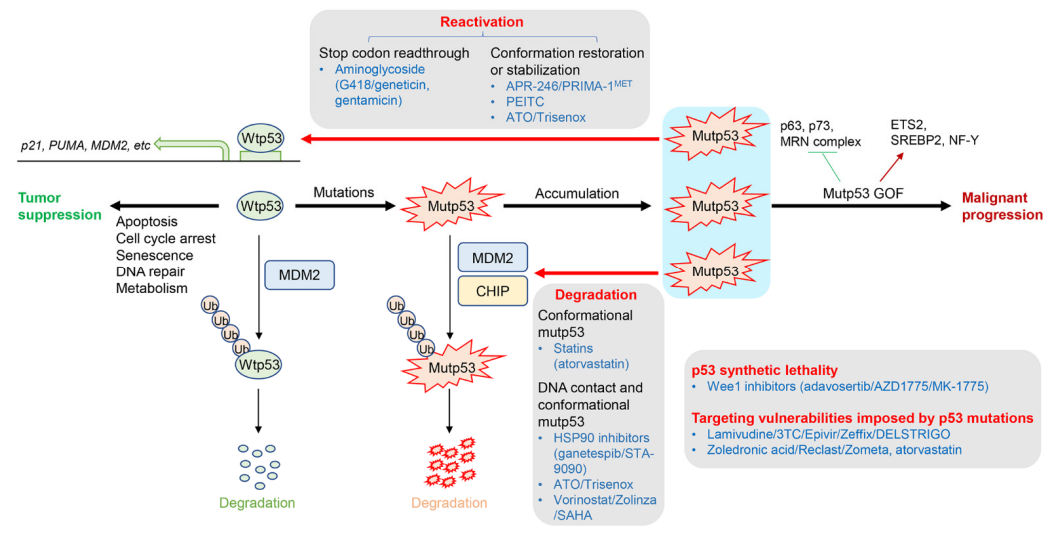
p53是一种转录因子,可调节参与细胞凋亡、细胞周期停滞、衰老、DNA修复和细胞代谢的许多下游靶基因的转录,从而起到肿瘤抑制作用[2]。
蛋白质水平和活性的wtp53在非应力条件下保持较低水平,主要是通过MDM2降解。
在遗传毒性条件下,wtp53通过磷酸化或乙酰化的翻译后修饰(PTM)稳定和激活,以诱导细胞周期停滞和/或细胞死亡(图1)[3]。
一旦wtp53功能因突变或缺失而受损,细胞就会失去对其生长的控制,从而促进肿瘤发生。越来越多的证据表明,p53是人类癌症中最常见的突变基因,在超过50%的人类癌症中发生突变。大多数p53突变是DNA结合域中的错义突变。
p53突变导致作为转录因子和肿瘤抑制因子的功能丧失(LOS)。错义mutp53 经常在肿瘤中积聚,以独立于wtp53的方式促进恶性进展、转移和耐药性。这些致癌性mutp53活性被称为功能获得(GOF)(图1)。
mutp53 GOF的机理主要是由mutp53与肿瘤抑制因子结合的能力引起的(例如,p63,p73,MRN复合物)和癌基因(例如,ETS2,SREBP2,NF-Y)以改变这些结合伴侣的功能。
临床上,肿瘤中mutp53的存在与多种类型癌症患者的晚期临床分期、转移和不良结局密切相关。鉴于p53的突变通常在肿瘤中特异性观察到,并且在非肿瘤组织中很少见,因此mutp53是理想用于癌症治疗的治疗靶点。

靶向p53突变的策略
1
直接靶向错义mutp53,以恢复wtp53的活性或稳定wtp53构象
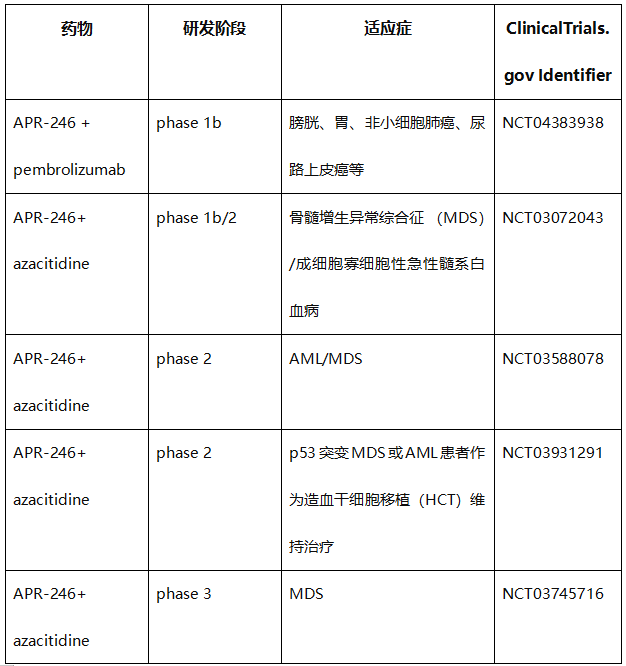
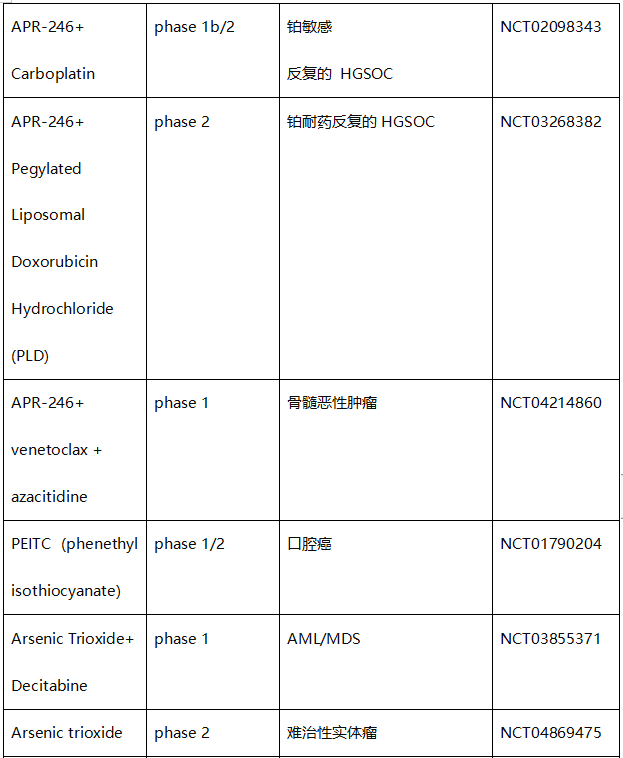
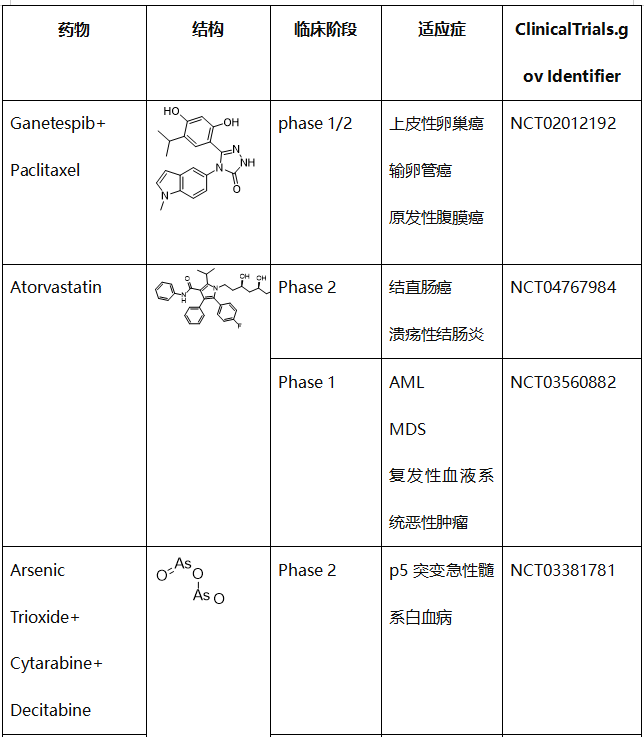
表1. 直接靶向错义mutp53的化合物的临床试验

2
mutp53蛋白的消耗或降解
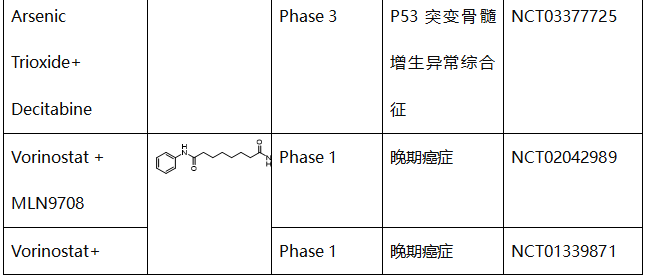
表2. 使mutp53蛋白的消耗或降解的化合物的临床试验

3
诱导p53合成致死性或靶向p53突变或缺失而导致增加的酶
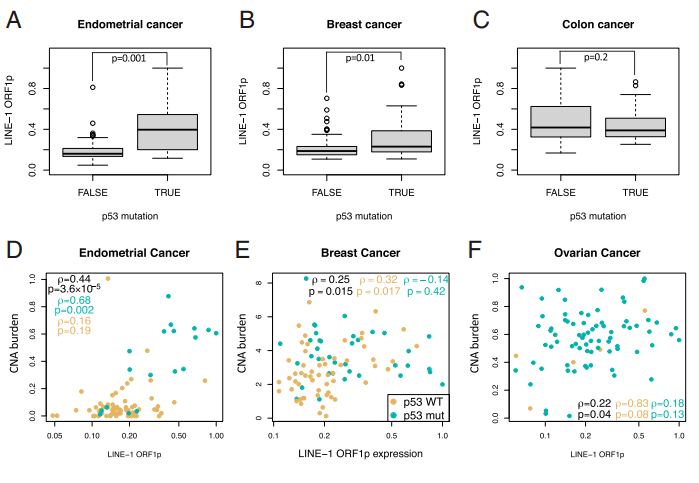
图2. A-C. 分别比较子宫内膜、乳腺癌和结肠肿瘤中 p53 野生型与突变肿瘤中 LINE-1 表达的箱形图;D–F. 子宫内膜癌、乳腺癌和卵巢癌中 LINE-1 表达与 CNA 负荷之间的 Spearman 相关性
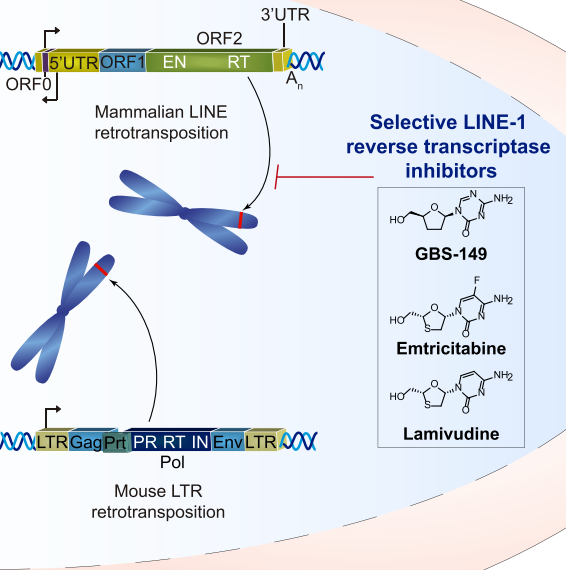
图3. Lamivudine抑制LINE-1
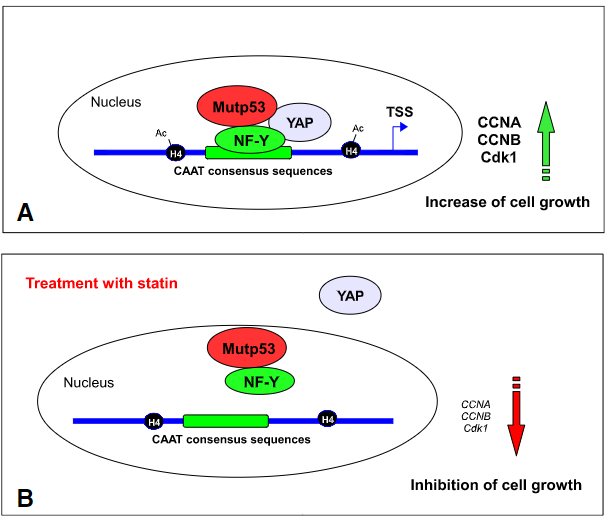
图4. mutp53与YAP/TAZ在细胞中作用
参考文献:(上下滑动查看更多)
1.Zambetti, G.P.; Levine, A.J. A comparison of the biological activities of wild-type and mutant p53. FASEB J. 1993, 7, 855–865.
2.Lane, D.; Levine, A. p53 Research: The Past Thirty Years and the Next Thirty Years. Cold Spring Harb. Perspect. Biol. 2010, 2, 000893
3.Shigeto Nishikawa and Tomoo Iwakuma, Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials, Cancers 2023, 15, 429.
4.Bykov, V.J.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K.G. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs. J. Biol. Chem. 2005, 280, 30384–30391.
5.Liu, W.-L.; Midgley, C.; Stephen, C.; Saville, M.; Lane, D.P. Biological significance of a small highly conserved region in the N terminus of the p53 tumour suppressor protein. J. Mol. Biol. 2001, 313, 711–731.
6.Wilson McKerrow, Xuya Wang, Carlos Mendez-Dorantes, Paolo Mita, Song Cao, Mark Grivainis, Li Ding, John LaCava, Kathleen H. Burns, Jef D. Boeke, and David Fenyo, LINE-1 expression in cancer correlates with p53 mutation, copy number alteration, and S phase checkpoint, PNAS 2022 Vol. 119 No. 8 e2115999119.
7.Guillermo Banuelos-Sanchez, LauraSanchez, Maria Benitez-Guijarro, ..., Francisco Franco-Montalban, Juan A. Tamayo, Jose L. Garcia-Perez, Synthesis and Characterization of Specific Reverse Transcriptase Inhibitors for Mammalian LINE-1 Retrotransposons, Cell Chemical Biology 26, 1095–1109.
8.Di Agostino, S.; Sorrentino, G.; Ingallina, E.; Valenti, F.; Ferraiuolo, M.; Bicciato, S.; Piazza, S.; Strano, S.; Del Sal, G.; Blandino, G. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. 2016, 17, 188–201.
只有化学团队,没有模型?
自建生物团队,成本太高?
“肿瘤药物研究合作”
(备注:合作)

服务:
本公众号免费接受科研团队/单位的研究进展、研发故事等非商业/非盈利目的投稿,及免费发布科研团队的招聘广告等,欢迎后台联系。
声明:发表/转载本文仅仅是出于传播信息的需要,并不意味着代表本公众号观点或证实其内容的真实性。据此内容作出的任何判断,后果自负。若有侵权,告知必删!
长按关注本公众号


本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言