靶点:PI3K
2022-12-21 精准药物 精准药物 发表于上海

PI3K是一种胞内磷脂酰肌醇激酶,与Src和RAS等癌基因的产物相关。PI3K信号通路是人类癌症中最常被激活的信号通路之一,几乎介导50%恶性肿瘤的发生。
PI3K是一种胞内磷脂酰肌醇激酶,与Src和RAS等癌基因的产物相关。PI3K信号通路是人类癌症中最常被激活的信号通路之一,几乎介导50%恶性肿瘤的发生。研究表明,PI3K信号通路的过度活跃与肿瘤的进展、肿瘤微血管密度的增加、癌细胞的趋化性和侵袭性增强有显著的相关性。目前,PI3K已成为治疗各种恶性肿瘤的重要靶点,包括乳腺癌和滤泡性淋巴瘤等。近年来,针对PI3K的选择性抑制剂已经被成功开发,目前全球总共批准5款PI3K抑制剂。然而由于毒性问题未能有效解决,PI3K抑制剂的研发遭遇了诸多挫折。
PI3K靶点介绍
磷酸肌醇3-激酶(PI3Ks)信号位于酪氨酸激酶、G蛋白偶联受体(GPCRs)和GTP酶(如:RAS、RAC、CDC42)的下游,以调节一系列细胞活动,包括代谢、增殖和迁移(图1)。PI3K信号通路是癌症中最常见的异常激活通路之一,早期研究表明,泛PI3K抑制剂LY294002可以恢复癌细胞对常规治疗的耐药性,包括化疗、放疗和靶向治疗。此外,一些PI3K家族成员也参与了炎症和自身免疫。
基于底物特异性和序列同源性,目前已经鉴定出三种不同类型的PI3K,它们由不同的酶亚型组成,在细胞信号转导过程中发挥特定的功能。在三个不同的组中,I类PI3K的活性似乎在肿瘤发生过程中至关重要。这类可分为两个亚家族:Ia类PI3K,依次分为三种不同的亚型PI3Kα, β和δ,由生长因子受体酪氨酸激酶(RTK)激活;Ib类PI3K(PI3Kγ亚型)被G蛋白偶联受体(GPCRs)激活。PI3K的催化亚基也被称为p110。
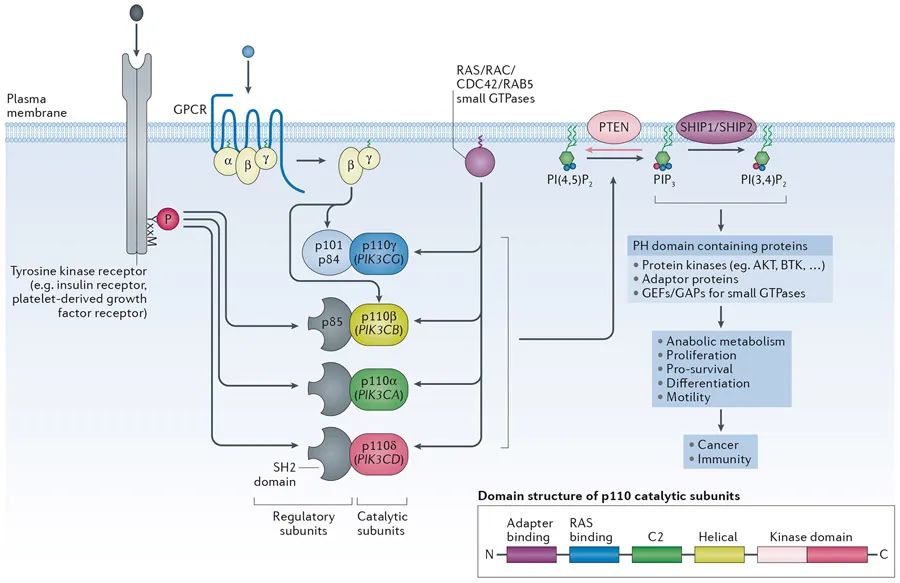
图1.PI3K结构及其介导的信号通路
PI3K抑制剂种类
研究表明,抑制PI3K被认为是一种有前景的策略,可用于治疗各种癌症,包括卵巢癌、乳腺癌、前列腺癌、胃癌、结直肠癌、胶质母细胞瘤、子宫内膜癌和脑癌等。
根据其选择性,目前鉴定的PI3K抑制剂可分为三大类:
1.泛PI3K抑制剂
2.PI3K异构体选择性抑制剂
3.双重抑制剂
泛PI3K抑制剂
Pan-PI3K抑制剂是三磷酸腺苷(ATP)竞争性抑制剂,针对所有四种I类亚型,因此它们的同时阻断是限制癌细胞增殖和存活的有效方法(图2)。然而泛抑制剂缺乏选择性,造成整个通路的非选择性抑制,从而导致严重的不良副作用。
Wortmannin和LY294002是第一代PI3K的非特异性抑制剂。研究发现,Wortmannin是一种已知的真菌类固醇代谢产物,1993年被鉴定为PI3K抑制剂。由于它与ATP结合位点上保守的赖氨酸残基形成强烈的共价键,诱导蛋白的大量构象畸变,以及几个额外的非共价相互作用,它对所有PI3K异构体产生强烈的不可逆抑制。但它在选择性和稳定性方面存在许多缺陷。
LY294002是第一个合成的PI3K抑制剂,它是槲皮素的类似物。槲皮素是一种天然类黄酮,以前被研究为PI3K抑制剂,其IC50在μM范围内。LY294002对PI3Kα、β、γ和δ亚型的IC50值分别为0.55、16、12和1.6 μM,是槲皮素的2.7倍。但其溶解度差、生物利用度低和代谢降解快,无法进一步的体内实验。
buparisib是一种有效的口服生物可利用的I类泛PI3K抑制剂,对PI3Kα, β, γ和δ亚型有效,IC50分别为52, 166, 262和116 nM。buparisib已被证明能够抑制癌细胞增殖和血管生成,目前正在进行临床试验。
Copanlisib(BAY80-6946,上市名称为Aliqopa),这是一种有效的泛PI3K药物(对PI3Kα,β,γ和δ的IC50分别为0.5,3.7,0.7和6.4 nM),于2017年获得批准上市。Pilaralisib是一种在nM浓度下活性的可逆抑制剂,并在患者中通过了II期临床研究晚期或复发性子宫内膜癌。
Taselisib(GDC-0032)对PI3K四种亚型都有较强的抑制作用,但对PI3Kα具有明显更高的效力。在临床前研究中,发现该化合物在多种类型的PIK3CA基因突变的癌症中具有活性,包括子宫浆液性癌、头颈部鳞状癌和乳腺癌。
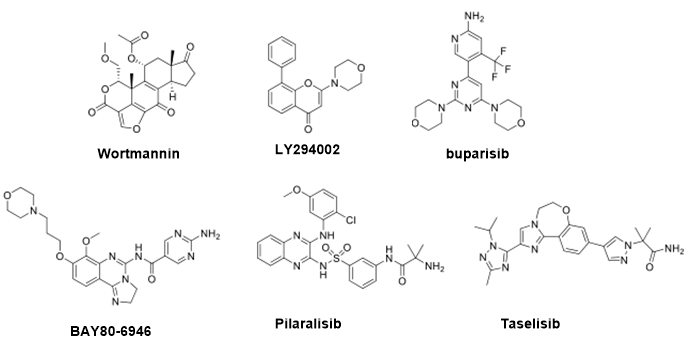
图2.泛PI3K抑制剂结构
PI3Kα, β, γ和δ亚型选择性抑制剂
为了限制包括免疫抑制和葡萄糖耐受不良在内的毒性作用的出现,研究人员针对每种I类PI3K亚型开发了选择性ATP竞争性抑制剂。
1)PI3Kα选择性抑制剂
I类PI3K催化亚基(PIK3CA)的α亚型,最常被编码该蛋白的基因分子结构的改变而激活。近年来,已经发现了几种能够选择性抑制这一子类的抑制剂(图3)。其中一些进入了临床试验,一些获得了FDA的批准。
Serabelisib(MLN1117, INK1117, TAK-117)是第一个被发现的口服生物可用PI3K抑制剂,对PI3Kα的选择性是其他亚型的100倍。在对携带PIK3CA基因突变的肿瘤细胞系进行的体外研究中,Serabelisib通过阻断细胞增殖和凋亡对PI3K通路产生了有效的抑制作用,而在PTEN基因突变的癌症模型中发现它无效。
Inavolisib(GDC-0077)被认为是PI3Kα的高选择性抑制剂。口服后证明对HR+/ her2乳腺癌有效,并进入III期临床试验。与其他PI3Kα抑制剂不同,Inavolisib阻碍突变的PI3K信号通路和细胞活力,其机制导致突变p110α蛋白降解,而不会对野生型p110α蛋白造成显著损伤。这种特殊的特性使该药物在PIK3CA基因突变的癌症中具有更好的治疗指标。从I期和II期临床试验中获得的结果表明,Inavolisib在伴有PIK3CA突变的HR+/HER2-(激素受体阳性/人表皮生长因子受体2阴性)乳腺癌患者中,与其他化疗药物联合使用时具有更高的抗癌活性和更高的安全性。然而,使用Inavolisib会引起一系列不良反应,包括高血糖、腹泻、皮疹和血小板减少。目前,该药与Palbociclib和Fulvestrant联合,正在晚期乳腺癌患者中进行III期临床试验。
Alpelisib也是一款口服PI3K抑制剂,它被证明对PIK3CA基因突变表达的PI3Kα亚型具有很强的选择性。最近,Alpelisib已被FDA批准与mTOR抑制剂Everolimus联合治疗内分泌肿瘤。
选择性PI3Kα抑制剂CH5132799是通过对肌球蛋白轻链磷酸化和血小板活化的研究发现的。它对PI3Kα亚型的IC50 = 14 nM,并被提出用于几种形式的腺癌的治疗。最近,该药物正在进行I期临床试验,用于晚期实体瘤患者的口服给药。

图3. PI3Kα选择性抑制剂
2)PI3Kβ选择性抑制剂
尽管PI3Kβ亚型与PI3Kα在结构和功能上有明显的相似之处,但在与特定底物的相互作用和功能方面存在实质性差异。到目前为止,已经确定了几种能够选择性靶向I类PI3K催化亚基β亚型的化合物,该亚基通常在PTEN抑制功能丧失后被激活(图4)。
PI3Kβ选择性抑制剂的第一个例子是TGX- 221,此前的研究发现它可以作为抗血栓和抗肿瘤药物。由于其手性性质,r -异构体TGX-221-R在抑制PI3Kβ作用方面比其他对映体活性高100倍。
GSK2636771对β亚型具有最佳效力和选择性,在口服PTEN缺陷晚期肿瘤患者中进行了开放标签I/IIa期剂量递增研究。
SAR260301表现出良好的药代动力学特性(口服生物利用度70%;T1/2 = 6.9小时(雌性裸大鼠))。相比于β亚型及其他激酶具有70倍的选择性。目前,SAR260301在晚期癌症患者的I/Ib期临床试验已经完成。尽管具有可接受的安全性,但该候选药物的临床开发因其快速的消除率而中断,导致较低的药物暴露水平。

图4. PI3Kβ选择性抑制剂
3)PI3Kγ选择性抑制剂
PI3Kγ抑制剂是能够选择性抑制I类PI3Kγ亚型的化合物,PI3Kγ在白细胞中表达,在趋化因子依赖性白细胞的趋化性和肥大细胞的激活中起关键作用。这种类型的药物在治疗各种疾病,如炎症、呼吸和代谢疾病以及癌症方面非常有用。然而,由于不同的PI3K亚型之间具有很强的结构相似性,迄今为止,很少有PI3Kγ亚型的真正选择性抑制剂被确定(图5)。
喹唑啉-噻唑烷衍生物AS605240是第一个选择性PI3Kγ atp竞争性抑制剂(IC50 = 8 nM),也具有良好的细胞渗透性,可用于治疗阿尔茨海默病。该化合物目前正处于I期临床试验阶段,用于联合治疗局部晚期或转移性实体瘤。
研究人员通过高通量化学蛋白质组学研究发现三唑吡啶衍生物的PI3Kγ抑制剂CZC24832,其在体外和体内炎症模型中都表现出较好的治疗效果。进一步的研究表明,该化合物作为一种有效的选择性PI3Kγ抑制剂(IC50 = 27 nM),对PI3Kβ具有10倍的选择性,对PI3Kα和PI3Kδ具有100倍的选择性。
在过去的十年中,研究人员通过对异喹啉类化合物的构效关系研究发现了Eganelisib(IPI-549),Eganelisib是一种高选择性的PI3Kγ抑制剂(IC50 = 0.29 nM),该药物用于多种癌症形式的口服免疫肿瘤治疗,具有调节抗肿瘤免疫反应的能力。
化合物AZD3458是另一种有效的特异性PI3Kγ抑制剂,它对催化亚基具有选择性抑制作用,IC50值为7.9 nM,与其他催化亚基相比具有100倍的选择性。临床前结果表明,该化合物在提高抗肿瘤免疫反应方面有显著效果。
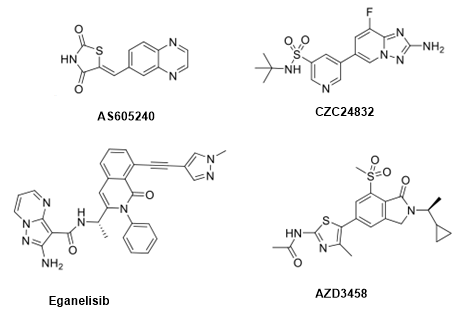
图5.选择性PI3Kγ抑制剂
4)PI3Kδ选择性抑制剂
I类PI3K催化亚基δ亚型(PI3Kδ)是治疗血液系统恶性肿瘤和免疫疾病的一个有前景的靶点,因为它在B细胞、T淋巴细胞、肥大细胞和中性粒细胞的PI3K/PKB/mTOR信号通路中起着至关重要的作用(图6)。
第一类PI3Kδ特异性抑制剂,又称为螺旋桨形抑制剂。Idelalisib(CAL-101,作为Zydelig上市)是第一种有效的口服选择性抑制剂(IC50 = 2.5 nM)。目前,该药已被批准用于慢性淋巴细胞白血病、滤泡性淋巴瘤和小淋巴细胞淋巴瘤的治疗。由于出现严重的腹泻、结肠炎甚至致命的肝毒性等严重副作用,该药物的临床应用受到限制。研究人员进一步优化得到的GNE-293具有改进的药代动力学特征(Ki = 0.47 nM,对PI3Kδ的选择性是PI3Kα的256倍),且没有遗传毒性作用。
第二代PI3Kδ抑制剂具有不同的作用机制。这类抑制剂以5,6,7,8-四氢吡啶[4,3-d]嘧啶衍生物Leniolisib(CDZ173)为代表,与第一代抑制剂相比,它表现出优越的安全性。在成功通过毒理学研究和I期临床试验后,Leniolisib已经完成了活化PI3Kδ综合征和原发性Sj¨ogren’s综合征的临床试验。此外,该药物已在一项开放标签、非随机扩展研究中进行,以评估长期安全性、耐受性、疗效和药代动力学。
Nemiralisib(GSK-2292767)是另一种非螺旋桨型PI3Kδ抑制剂,已在哮喘和慢性阻塞性肺疾病治疗的II期临床试验中进行了测试。该化合物在治疗由衰老T细胞引起的活化PI3Kδ综合征方面表现出积极作用。在细胞中,该药物已显示出足够的效力和优良的选择性。
Umbralisib(Ukoniq, TGR1202)是一种具有口服活性的新一代特异性和安全的PI3Kδ抑制剂(IC50 = 22 nM)。据观察,该化合物对酪蛋白激酶-1ε也有一定的抑制活性,酪蛋白激酶-1ε是一种对调节T淋巴细胞功能具有抑制活性的蛋白质。因此,它可以被认为是一种双重PI3Kδ/CK-1ε抑制剂。最近,Umbralisib已被批准作为治疗成人复发或难治性边缘区淋巴瘤的二线药物和治疗复发或难治性滤泡性淋巴瘤的三线药物。目前正在进行治疗各种血液恶性肿瘤的临床试验。
第二代PI3Kδ抑制剂的另一个例子是Acalisib(GS-9820)。这种药物被提出可用于治疗一些肿瘤,如慢性淋巴白血病(IC50 = 12.7 nM)。

图6. PI3Kδ选择性抑制剂
PI3K异构体双重抑制剂
目前批准的用于治疗复杂、异质和多基因疾病的药物大多数基于“单靶点/单药物”模式。由于这些单一活性成分的药物通常效果较弱,因此为了获得更大的治疗效果,联合使用是一种有效的策略。然而,这种联用并不总是能带来预期的好处,因为药物不良相互作用、低安全性、不可预测的药代动力学和患者“依从性”差往往会阻碍它们。为了克服这些缺陷,研究人员的兴趣转向了多靶点药物的发现和开发(图7)。
在这些新一代药物中,口服活性Pictilisib(GDC0941)是双PI3Kα/δ抑制剂最重要的例子之一(两种亚型的IC50 = 3.0 nM)。在II期临床研究中,Pictilisib与其他抗癌药物联合治疗乳腺癌、晚期实体瘤和非鳞状非小细胞肺癌显示出良好的安全性。
Duvelisib (IPI-145,上市名为Copiktra)是一种异喹啉酮衍生物,被证明能够抑制两种异构体,IC50分别为50和0.24 nM,对δ异构体的抑制能力高于特定的PI3Kδ抑制剂。临床上,Duvelisib被批准用于治疗血液系统恶性肿瘤,如慢性淋巴细胞白血病,小淋巴细胞淋巴瘤和滤泡性淋巴瘤。
新一代PI3Kγ/δ抑制剂家族的另一个成员是Tenalisib(RP6530)。PI3Kγ和PI3Kδ两种亚型的IC50分别为33.2和24.5 nM。临床前研究表明,Tenalisib能够诱导凋亡和抗增殖活性,从而减少血管生成。这种化合物目前正在进行II期研究,用于治疗不同类型的癌症。

图7.PI3K异构体双重抑制剂
PI3K抑制剂研究现状
已上市的PI3K药物
PI3K抑制剂种类很多,根据作用机制,大致可分为广谱型PI3K抑制剂(pan-PI3K)、亚型特异性PI3K抑制剂和靶向PI3K/mTOR双重抑制剂三大类。据统计,全球总共批准5款PI3K抑制剂(图8),即吉利德的idelalisib、拜耳的copanlisib(Zydelig)、拜耳的Copanlisib(Aliqopa)、Verastem公司的duvelisib(Copiktra)、诺华的alpelisib(BYL719,Piqray)和TG Therapeutics的Umbralisib。

图8 . 已上市的PI3K抑制剂
PI3K抑制剂的临床研究汇总
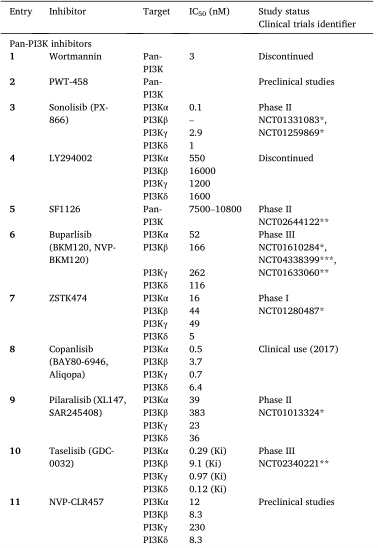
图9. 处于临床阶段的泛PI3K抑制剂

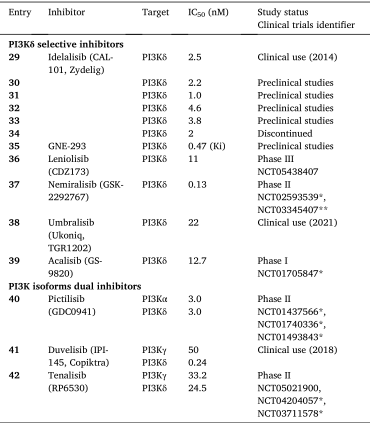
图10.处于临床阶段的PI3K选择性抑制剂
— 小结 —
多项研究均指出PI3K是一个具有临床潜力的抗癌靶点,并且已经成功推动了5款PI3K药物上市。但目前,针对PI3K抑制剂的药物选择性、耐药性以及安全性问题,却始终没有一个完美的“参考答案”。其中,安全性问题是PI3K抑制剂的首要解决问题。期待更多安全有效的PI3K抑制剂获批上市,造福肿瘤相关患者。
参考文献:
1、Recent advances in PI3K/PKB/mTOR inhibitors as new anticancer agents. European Journal of Medicinal Chemistry 246 (2023) 114971
2、PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Front. Oncol. 12 (2022), 819128 https://doi.org/10.3389/fonc.2022.819128.
3、PI3K inhibitors are finally coming of age. doi.org/10.1038/s41573-021-00209-1
4、The PI3K pathway in human disease. Cell 170, 605–635 (2017).
5、Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988–1004 (2005).
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言