靶向mTOR抑制剂研究现状及未来方向
2023-01-03 精准药物 精准药物 发表于上海

雷帕霉素靶蛋白mTOR是一种丝氨酸/苏氨酸激酶 , 因其是雷帕霉素的作用靶点而得名。以mTOR为核心的mTOR复合体(mTORC)是许多重要细胞信号通路的核心.
哺乳动物雷帕霉素靶蛋白mTOR是一种丝氨酸/苏氨酸激酶 , 因其是雷帕霉素的作用靶点而得名。以mTOR为核心的mTOR复合体(mTORC)是许多重要细胞信号通路的核心 , 因此mTOR在维持细胞能量代谢平衡、影响细胞分裂、参与细胞自噬和凋亡等方面有着重要作用。
研究发现,mTOR信号的失调与mTOR相关疾病的并发症有关,并且在不同类型的癌症中都观察到过度激活的mTOR信号。此外,mTOR与阿尔茨海默病、帕金森病、亨廷顿病等神经退行性疾病以及糖尿病、肥胖和衰老密切相关。因此,抑制mTOR信号通路可有效阻断各种生长因子的异常信号转导,从而阻断疾病的发生和发展。目前,针对mTOR靶点的药物开发也是一个热门赛道,并且已经成功开出了三代mTOR抑制剂。

mTOR结构及介导的信号通路
mTOR是大小为289 kDa丝氨酸/苏氨酸蛋白激酶。由于其催化结构域表现出与脂质激酶(如PI3K)相似的结构特征,因此它被纳入磷脂酰肌醇-3激酶相关激酶(PIKK)家族中。mTOR激酶由mTOR基因编码,能够结合不同的蛋白质形成两种类型的多蛋白复合物:由5个亚基(mTOR、mLST8、RAPTOR、PRAS40和DEPTOR)组成的mTOR复合物1 (mTORC1)和由6个亚基(mTOR、DEPTOR、mLST8、RICTOR、mSIN1和PROTOR)组成的mTORC2(图1)。
虽然此前的研究表明只有mTORC1对雷帕霉素敏感,雷帕霉素是一种通过与其FKBP -雷帕霉素结合(FRB)结构域相互作用而结合mTOR的天然产物。但随后的研究发现,长期使用抗生素治疗也会抑制mTORC2活性。mTORC1主要负责调节细胞生长机制,而mTORC2有助于控制细胞生存和增殖。
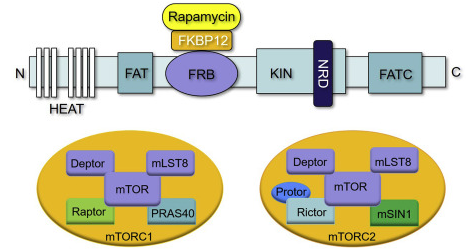
图1.mTOR及其复合物的结构
研究表明,mTOR通路受多种细胞信号的调控,包括有丝分裂生长因子、胰岛素等激素、营养素(氨基酸、葡萄糖)、细胞能量水平和应激条件。
其中,PI3K/Akt信号通路是通过mTOR传递信号的主要通路,在介导细胞存活和增殖中起重要作用。PI3K/Akt通路的信号是由与细胞膜上的受体结合的生长因子的有丝分裂刺激启动的,这些受体包括IGFR(胰岛素样生长因子受体)、PDGFR(血小板衍生生长因子受体)、EGFR(表皮生长因子受体)和HER家族。来自激活的受体的信号直接传递到PI3K/Akt通路,或者也可以通过由致癌蛋白RAS激活的生长因子受体激活。此外,AKT也可以被PDK-1(磷脂依赖激酶-1)磷酸化和激活,AKT可以直接磷酸化mTOR。AKT也可能通过TSC1/TSC2(结节性硬化症复合体)的作用间接作用于mTOR。也有研究表明,磷脂酸PA也能激活mTOR信号通路。
此外,mTOR的激活会导致下游靶点的磷酸化。除了对翻译的影响外,mTOR还通过调节RNA聚合酶I和III来调节蛋白质的合成,而这两个聚合酶负责核糖体和转运RNA的转录。
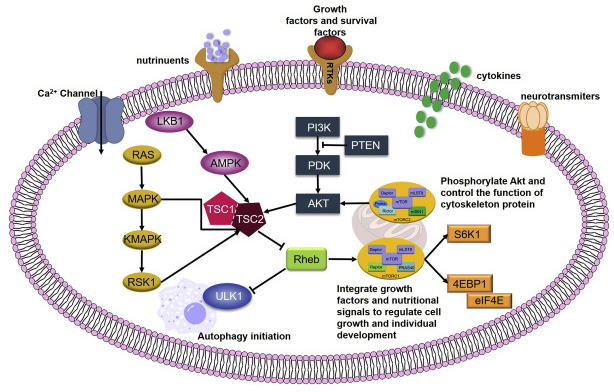
图2.mTOR介导的信号通路

mTOR抑制剂种类
mTOR是细胞内的一种基本关键蛋白,是汇聚和分类大量生物信息的关键节点,负责调节各种细胞生理过程。mTOR介导的通路在人类癌症中通常不受调控,因此为开发新的治疗药物提供了一个有吸引力的靶点。根据种类,mTOR抑制剂可分为三代:
1、抗生素类变构抑制剂(第一代)
2、ATP竞争抑制剂(第二代)
3、新型mTOR抑制剂(第三代)
1
抗生素类变构抑制剂
雷帕霉素(Rapamycin)及其衍生物,被称为Rapalogs,是mTOR的第一代变构抑制剂的成员,通过与肽基脯氨酰异构酶FKBP12形成复合物以介导抗增殖功能(图3)。FKBP12是一种普遍存在的蛋白质,可作为免疫抑制药物的受体 。
这些抑制剂的作用机制始于与FKBP12形成复合物,进而与mTOR FRB结构域结合,诱导构象变化,从而抑制mTORC1激酶活性。mTOR抑制意味着阻断蛋白质合成和细胞生长,并通过促进肿瘤消退来放大自噬过程。
研究发现,雷帕霉素是一种天然大环内酯类抗生素,从吸湿链霉菌污染的土壤样品中分离出来,具有抗真菌、抗增殖和免疫抑制特性。虽然雷帕霉素的IC50值为0.1 nM,生物半衰期为50 h。口服后,其不稳定性和水溶性差限制了其临床应用。为了提高生物利用度和其他药代动力学特性,几种雷帕霉素衍生物已经被开发出来,主要是针对母体化合物的42位的C原子进行改造。
第一个雷帕霉素衍生物是Temsirolimus,这是一种由雷帕霉素的42位OH与2,2-二羟甲基丙酸酯化得到的前药。该衍生物具有较好的稳定性和溶解性,作用时间较长(IC50 = 1.76 μM,半衰期17 h)。然而,Temsirolimus仅适用于静脉注射,并已被批准用于治疗晚期肾细胞癌,而在III期临床研究中,它被证明可以减缓套细胞淋巴瘤的进展。
一种口服活性雷帕霉素衍生物Everolimus是通过与乙二醇醚化而获得的,因此具有更好的稳定性和水溶性。Everolimus的IC50 = 5-6 nM,半衰期为30 h,已被批准用于治疗几种癌症,如晚期肾细胞癌、进行性胰腺源性神经内分泌肿瘤、胃肠道癌和肺癌。
另一种口服活性雷帕霉素衍生物Ridaforolimus是通过与二甲基膦酸酯化得到。然而,这种化合物只能延缓肿瘤的进展。在一项I期研究中,Ridaforolimus对患有晚期实体瘤的儿科患者具有良好的耐受性。
Umirolimus是一种高亲脂性的雷帕霉素衍生物,主要特征是血管壁和平滑肌细胞膜穿越率高。当在细胞内时,它会导致其生物周期的停止,并减少血管狭窄的进展。此外,Umirolimus表现出强大的免疫抑制和抗增殖活性。
研究人员进一步用四唑环取代雷帕霉素42位羟基得到Zotarolimus。由于其高亲脂性,这种化合物适用于载药包载递送,以保证足够长的释放时间。由于雷帕霉素结构的复杂性,通过化学合成其类似物被证明是不现实的。
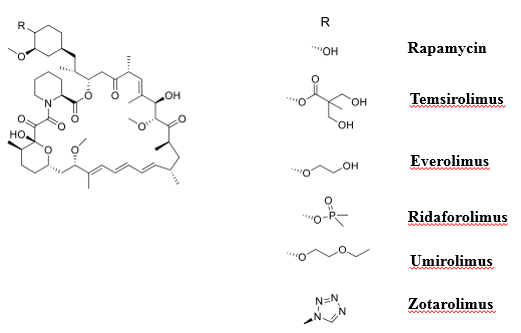
图3.雷帕霉素及其衍生物结构
2
ATP竞争性抑制剂
第二代mTOR抑制剂与第一代mTOR抑制剂结构具有很大的差别。第二代的mTOR抑制剂主要聚焦于开发mTOR的小分子抑制剂(图4)。这类抑制剂选择性靶向mTOR的活性激酶位点,因此可以充当ATP竞争性抑制剂。同时,这些分子也被称为选择性mTOR激酶抑制剂(TORKIs),可确保mTORC1和mTORC2的完全阻断,从而防止PKB磷酸化。
第一类衍生物都含有一个吡啶[2,3-d]嘧啶骨架,并可口服生物利用。该类化合物的代表性例子是化合物AZD8055和Vistusertib,其IC50值分别为0.8 nM和2.8 nM。这两种化合物都能抑制mTORC1和mTORC2,导致下游蛋白S6K1, 4E-BP1和PKB的浓度依赖性磷酸化阻滞。尽管AZD8055的潜力值得关注,但由于肝毒性,其治疗实体癌和淋巴瘤的临床实验已经终止。而Vistusertib毒性较小,针对多种类型癌症(包括多形性胶质母细胞瘤和胃肿瘤)的多项II期临床试验最近已完成。
另一种第二代抑制剂是以4-氨基吡唑并[3,4-d]嘧啶为基本骨架的化合物。这类抑制剂的第一个例子是以Torkinib(IC50 = 8 nM)为代表,对mTOR的选择性比其他PI3K激酶高10倍,对细胞增殖的抑制作用呈剂量依赖性。Torkinib对白血病、胃癌和结肠癌等多种癌症均表现出良好的抗癌活性。在铂耐药细胞中,PKB/mTOR信号通路过度调控,这种化合物可以使癌细胞对卡铂的作用重新敏感。此外,Torkinib还被证明可以有效地增强对放射治疗的反应,并恢复放射抵抗癌细胞的敏感性。
另一种基于4-氨基吡唑并[3,4-d]嘧啶 衍生物Sapanisertib,具有口服生物可用性,并显示出良好的临床应用药理特性。这种化合物能够阻断两种复合物mTORC1和mTORC2,目前已经进入了治疗肝细胞癌、胶质肉瘤、胶质母细胞瘤和其他实体肿瘤的临床试验。
另一个第二代mTOR抑制剂的例子是Torin1,它是一种基于苯并[H]1,6-萘吡啶-2-酮的特殊母核的化合物,目前已被证明可以选择性抑制mTORC1和mTORC2, IC50值分别为0.29和2 nM。但其稳定性差,口服生物利用度低,半衰期很短,阻碍了进一步的体内研究。
Torin1经过进一步化学简化后得到了Torin2。虽然其抑制作用相对于Torin1较低(对mTORC1和mTORC2的IC50 分别为37和25 nM),但Torin 2具有更大的稳定性和更长的半衰期,可以进一步对各种类型的癌症进行临床前实验,包括甲状腺、肝脏、结直肠癌、乳腺癌和白血病。
OSI-027是一种口服选择性mTOR抑制剂,具有咪唑[5,1-F]三嗪结构(IC50 = 4 nM)。该化合物可有效抑制胰腺导管腺癌细胞增殖,提高吉西他滨的治疗效果。它在晚期实体瘤患者中也显示出剂量依赖的活性。但在用药后观察到对肾脏的毒性作用。
Onatasertib是一种以3,4-二氢吡啶并[2,3-B]吡嗪-2(1H)-酮为母核的 第二代mTOR抑制剂, IC50= 16 nM,已被证明能有效抑制头颈癌肝细胞和鳞状细胞的细胞生长。
JR-AB2-011是一种2-亚氨基噻唑烷衍生物,能够通过结合RICTOR亚基抑制mTORC2复合物的形成(IC50 = 360 nM),但对mTORC1没有任何影响。该化合物目前正在进一步研究中,因为它已被发现在多形性胶质母细胞瘤中具有潜在活性,对其表现出显著的抗癌特性。

图4.第二代mTOR抑制剂结构
3
新型mTOR抑制剂
第三代mTOR抑制剂又被称为RapaLink(图5),主要将ATP竞争性抑制剂Sapanisertib通过不同类型的连接链连接到Rapalogs的大环核,从结构上看,这种设计类似于双重抑制剂的设计策略。这种混合药物的使用将克服基于Rapalog或ATP竞争性mTOR抑制剂的单药治疗后出现的耐药性。此外,多重活性还会提高药物的选择性和疗效。
RapaLink的第一个例子由RapaLink 1、2和3表示。它们都是通过将Sapanisertib的N-1原子与Rapamycin的O-42原子连接而得到的。分子对接模拟显示,RapaLink 1和2具有长度超过30个原子的连接链,从而能够同时结合两个位点,而短连接链的RapaLink 3却不能实现这样的双重相互作用。
第三代mTOR抑制剂包括其他具有不同结构特征的药物。一个典型的例子是Palomid 529,这是一种基于6H-苯并[c]苯并吡喃-6-酮结构的口服活性mTOR抑制剂。据报道,Palomid 529能够同时结合mTORC1和mTORC2,从而抑制复合物的组装或导致已经组装的复合物的分解。该药物已被证明在前列腺癌中非常有效,且对放射治疗和其他化疗有致敏作用。并且,由于Palomid 529具有出色的透过血脑屏障的能力,Palomid 529有望成为治疗脑肿瘤的合适候选药物。

图5.第三代mTOR抑制剂结构

mTOR抑制剂研究现状
目前只有少数mTOR抑制剂应用于临床(图6)。并且所有已批准的mTOR抑制剂都属于第一代mTOR抑制剂。雷帕霉素是首个被FDA批准市的mTOR抑制剂,目前适用于预防器官移植后出现的器官排斥反应,可单独使用或配合钙调神经磷酸酶抑制剂或皮质类固醇使用。近年来,受市场前景吸引,mTOR抑制剂成为医药企业研发的热点之一。目前全球多个国家的企业都在积极布局mTOR抑制剂市场,包括美国辉瑞公司、礼来公司、诺华公司以及德琪医药、江苏开拓药业等企业。此外,也有多款mTOR抑制剂获批开展临床。
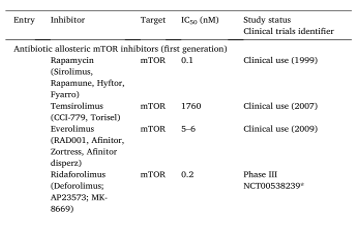

图6.已上市和处于临床阶段的mTOR抑制剂
— 小结 —
研究表明,mTOR激酶在致癌PI3K/AKT/mTOR信号通路调控中起关键作用,过度激活的mTOR功能与人类的几种病理状况有关。在过去的几十年里,人们已经开发出了多种mTOR抑制剂。目前的mTOR抑制剂分为三代,具有不同的作用机制,每一代抑制剂的抑制潜力也各不相同。其中,第三代mTOR抑制剂是通过Linker将雷帕霉素与ATP竞争性mTOR抑制剂连接起来,从而抑制耐药突变体,是重要的研究突破。
尽管变构mTOR抑制剂的发现最早,且目前对Rapalogs的研究已经相对成熟,但Rapalogs具有分子量大,结构复杂,合成难度较大,修饰位点有限等缺陷。因此,具有简化结构的第二代基于小分子mTOR抑制剂是接下来mTOR领域的重要研究方向。总的来说,mTOR有望成为下一个药物掘金之地。
参考文献:
1、Recent advances in PI3K/PKB/mTOR inhibitors as new anticancer agents. https://doi.org/10.1016/j.ejmech.2022.114971
2、Insights into the PI3-K-PKB-mTOR signalling pathway from small molecules, J. Chem. Biol. 1 (2008) 49-62,https://doi.org/10.1007/ s12154-008-0008-0.
3、Contemporary mTOR inhibitor scaffolds to diseases breakdown: A patent review (2015–2021)
4、Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle, J. Antibiot. (Tokyo) 28 (1975) 721–726
5、Inhibition of the immune response by rapamycin, a new antifungal antibiotic, Can. J. Physiol. Pharmacol. 55 (1977) 48–51, https://doi.org/10.1139/y77-007.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言