
《柳叶刀》子刊:胃肠道息肉病患者做基因检测有啥临床意义?
2023-12-17 苏州绘真医学 苏州绘真医学 发表于上海

本文综述了息肉病患者的已知综合征和基因、基因检测和临床管理,以及胃肠道息肉病领域的最新进展和挑战。
胃肠道息肉病的特征是存在多个息肉,尤其是在结直肠,包括癌症易感遗传综合征和非综合征性临床表现。在息肉病综合征中观察到的异质性与遗传原因、遗传方式、息肉负荷和组织学类型以及结肠外表现谱和频率有关。这些特征决定了携带者的临床管理,包括癌症预防和早期检测以及肿瘤治疗的策略。尽管在确定息肉病的遗传原因方面取得了重大进展,但很大一部分病例在遗传学上仍无法解释。虽然其中一些病例可能与生活方式、环境因素或癌症治疗相关,但可能会发现额外的息肉病易感基因。本文综述了息肉病患者的已知综合征和基因、基因检测和临床管理,以及胃肠道息肉病领域的最新进展和挑战。
研究背景
遗传性结直肠癌易感综合征是由遗传性癌症基因致病性变异引起的。无论是常染色体显性还是隐性遗传,这些综合征都可表现为息肉病或非息肉病表型(图 1)。经典基因型-表型相关性正在不断发展,揭示了一系列临床表现。

图1
林奇综合征是最常见的遗传性结直肠癌综合征,发生率约为 1/280-400。林奇综合征诊断不足,林奇综合征总人群中约有 5% 的病例被诊断出来。虽然林奇综合征不会引起经典的息肉病表型,并被归类为遗传性非息肉病性结直肠癌综合征,但患者可发展为多发性结直肠腺瘤。林奇综合征的定义是存在 DNA 错配修复基因 MLH1、MSH2、MSH6 或 PMS2 胚系杂合致病性变异或表观突变,这增加了结直肠错配修复缺陷癌症(子宫内膜癌、卵巢癌、胃癌和泌尿道肿瘤)的风险。
与错配修复缺陷的结直肠癌不同,家族性非息肉病性错配修复功能完整结直肠癌的遗传基础大多仍不明确。RPS20 胚系杂合致病性变异可引起罕见的遗传性非息肉病性结直肠癌综合征,特征是错配修复功能完整结直肠癌的风险高,没有结肠外表现。尽管其他分子诊断可能解释了遗传性非息肉病性结直肠癌的显性或隐性遗传病因,但迄今为止尚未确定分子或表型特征。
与遗传性非息肉病性结直肠癌相比,已知息肉病综合征的基因型和表型谱更为多样化(图 1)。这种多样性不仅由致病基因驱动,还由不同类型的息肉病(腺瘤性息肉病、错构瘤性息肉病、锯齿状息肉病或混合性息肉病)、遗传方式(显性或隐性)以及结肠外表型的存在和性质驱动(表 1)。本综述重点关注结直肠息肉病综合征,对已知的综合征和基因、肿瘤特征进行了遗传学和临床描述,并概述了基因检测和临床管理。
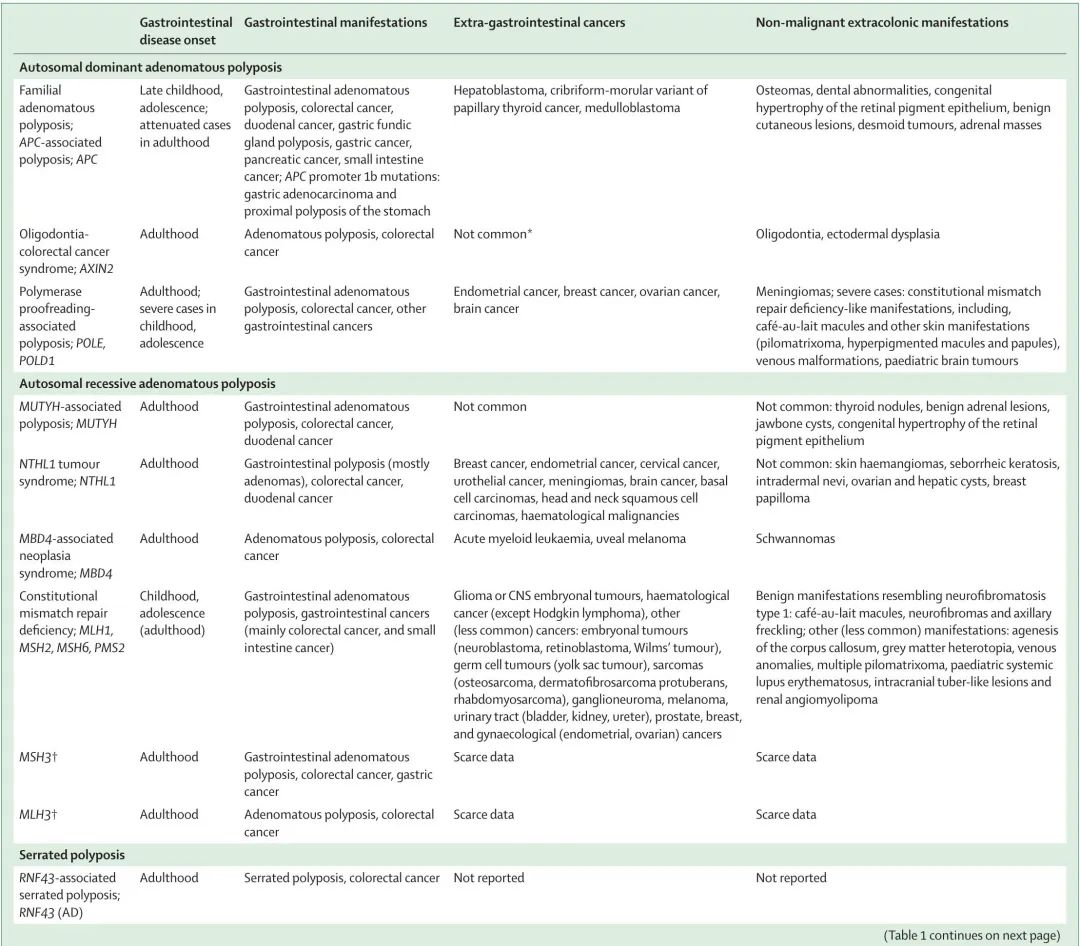
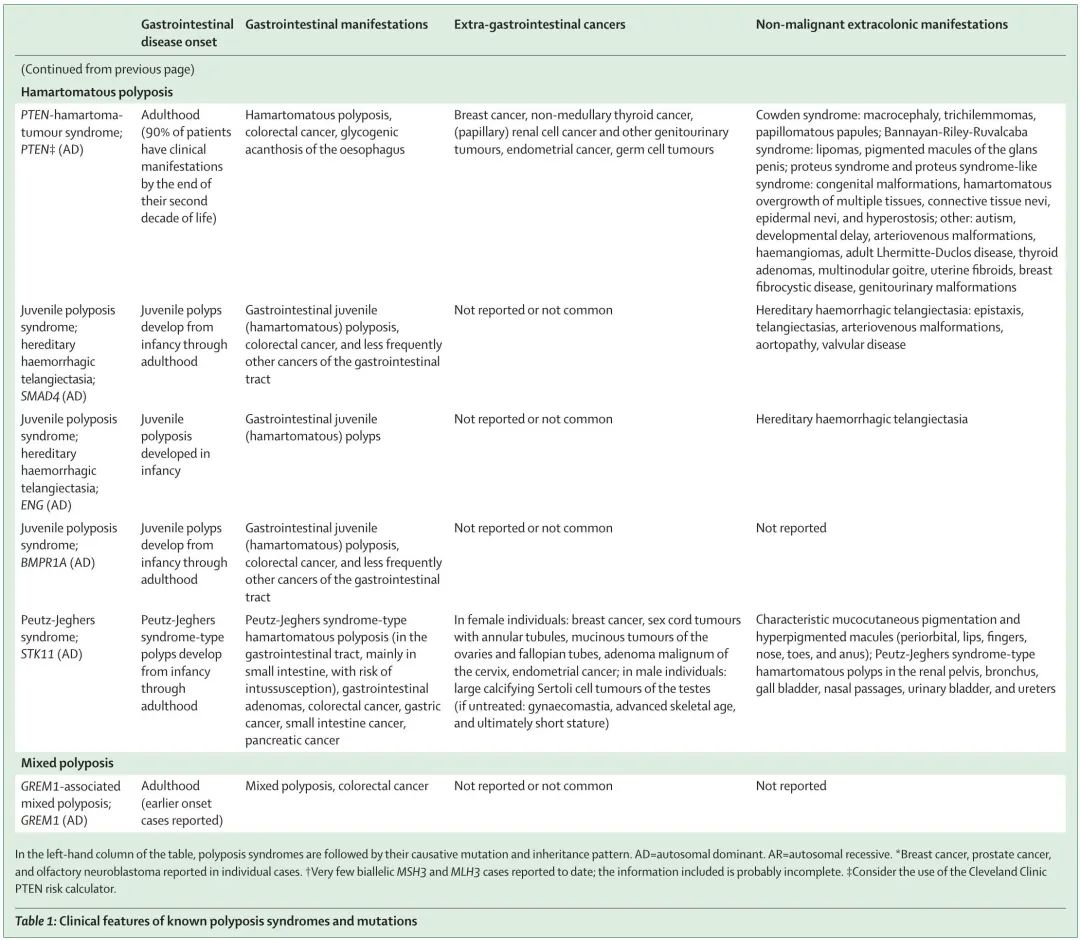
表1
息肉病综合征和致病基因
腺瘤性息肉病
常染色体显性遗传性腺瘤性息肉病综合征
息肉病综合征很少见,人群发病率低于 1/5000。鉴于这些综合征的广泛表型谱,其识别可能具有挑战性。家族性腺瘤性息肉病(FAP)的发病率约为 1/8500,是特征最明确和最常见的多发性腺瘤综合征。家族性腺瘤性息肉病的特征是在患者的一生中发现数百个结直肠腺瘤,通常在结直肠癌和息肉病家族史的背景下。这种情况还与结肠外特征有关,其中一些特征高度相关,特别是上消化道肿瘤和硬纤维瘤,这些是家族性腺瘤性息肉病相关死亡的主要原因。相关表型详见表 1,胃肠道临床表现见图 2 和图 3。家族性腺瘤性息肉病是由抑癌基因APC致病性胚系变异引起的。野生型等位基因体细胞失活导致APC完全失活,引起Wnt信号通路组成型激活,造成β-连环蛋白介导的癌基因和其他参与癌症相关过程的基因的激活。尽管家族性腺瘤性息肉病显示出常染色体显性遗传模式,大约 30% 的患者携带新发突变,没有该疾病家族史。
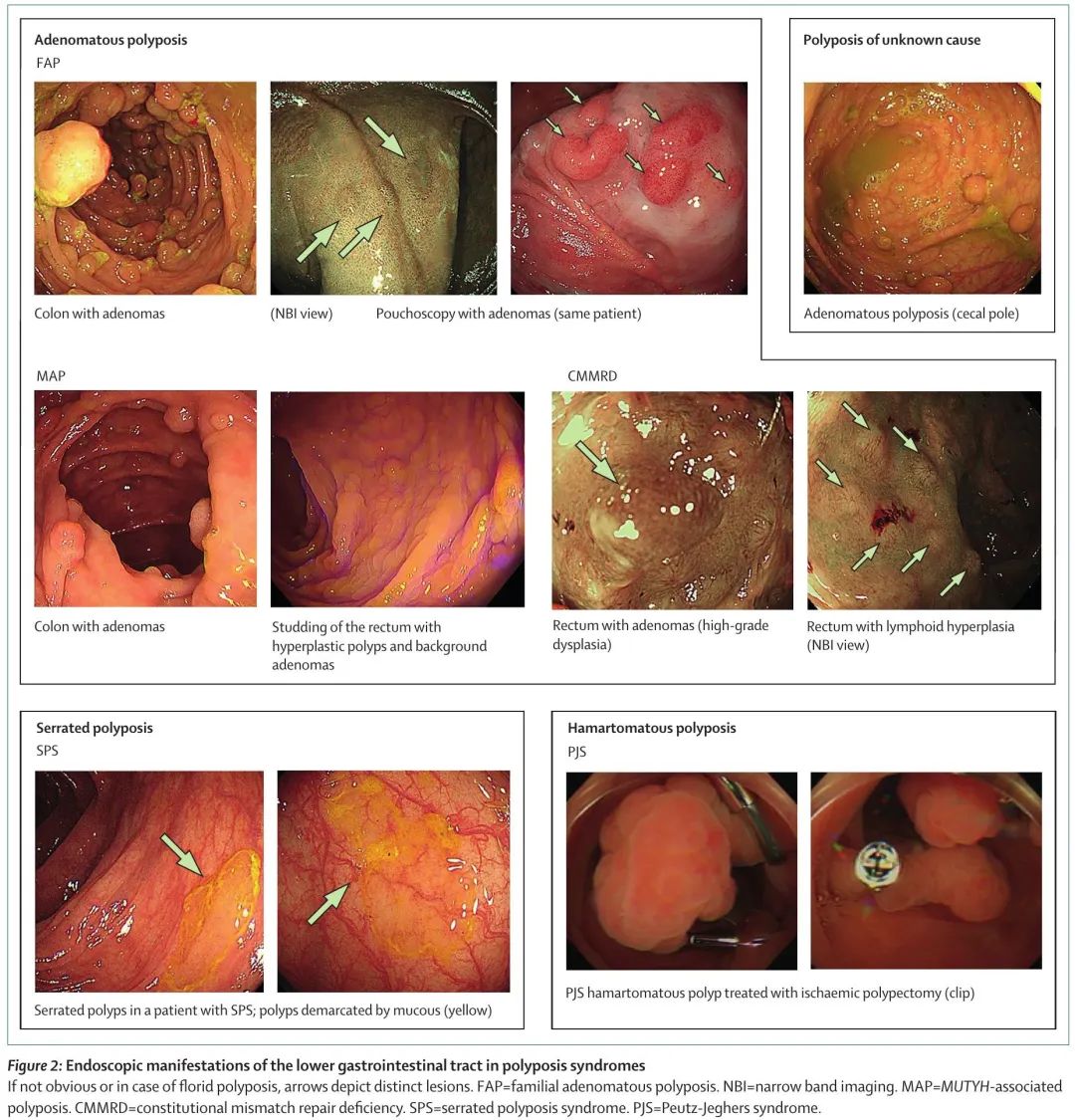
图2
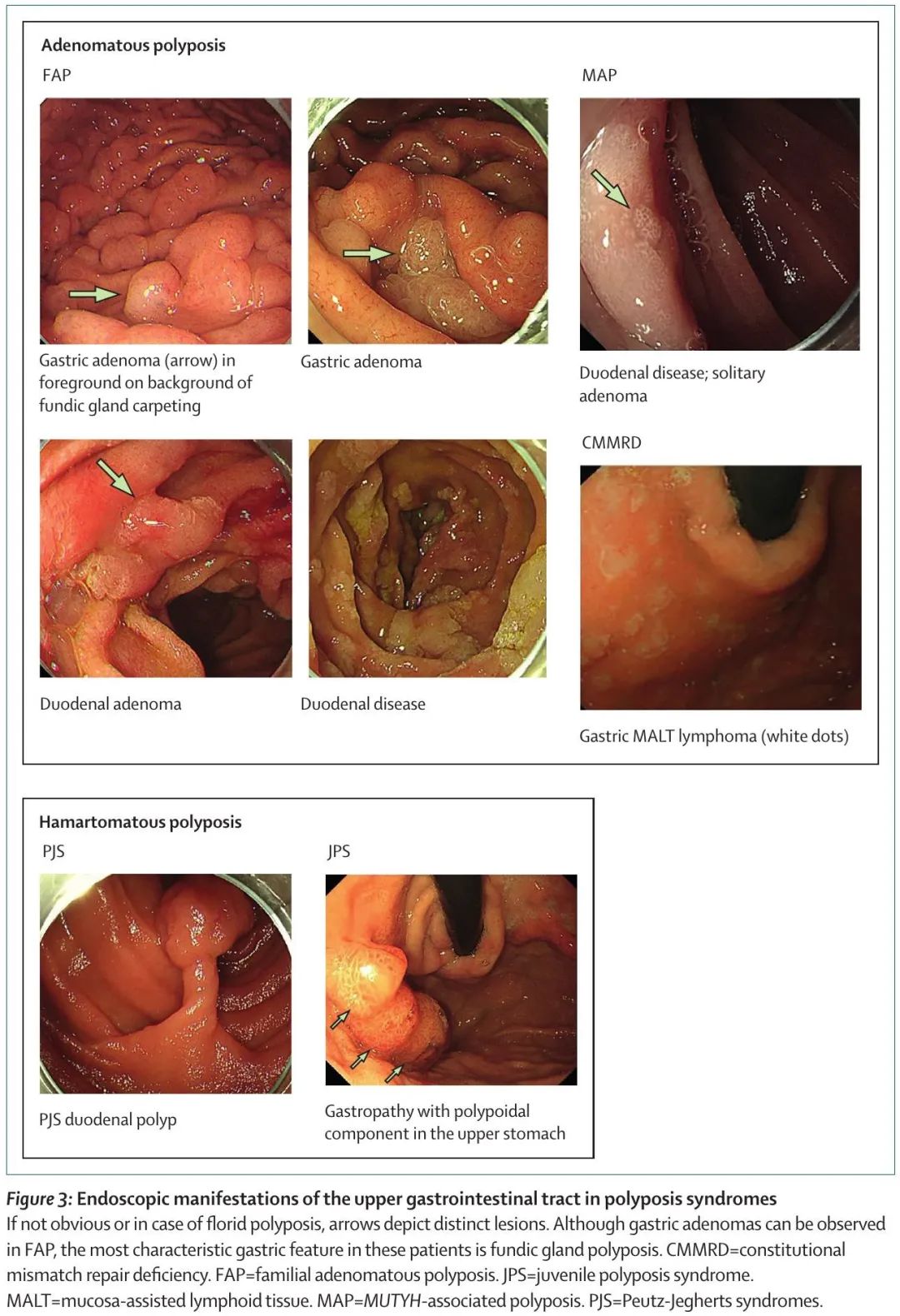
图3
APC基因变异位点可能决定了相关疾病的表型严重程度(基因型-表型相关性)。APC 基因外显子 15 包含一个突变簇区域,与严重表型相关,而 5ʹ 和 3ʹ 区域的致病性变异与较温和的表型相关,特征是存在少于 100 个结直肠腺瘤(这些较温和的表型统称为轻表型家族性腺瘤性息肉病)。一种称为胃腺癌和近端息肉病综合征的超罕见胃表型由 APC 外显子 1B(启动子)胚系突变引起。
家族性腺瘤性息肉病越来越多地被认为是一系列在临床表型上具有高度异质性的疾病,部分但不完全由基因型解释。因此,临床管理主要取决于上消化道和结肠镜检查,或息肉负荷,分布和癌症风险临床评估。
和 APC 一样,AXIN2 是一种 Wnt 阴性调节因子,AXIN2 胚系杂合致病性变异已在全球几例病例中发现,使个体易患与非典型牙列(少牙畸形)相关的多发腺瘤表型。
影响 DNA 聚合酶ε和δ校对功能的 POLE 和 POLD1 胚系致病性变异导致罕见的常染色体显性遗传性息肉病综合征,称为聚合酶校对相关息肉病。除了结直肠息肉病和癌症易感性外,这些变异杂合子携带者其他肿瘤类型的发病风险也增加,包括上消化道肿瘤、子宫内膜癌、乳腺癌、卵巢癌或脑肿瘤(表 1)。该综合征的临床表现通常出现在成年期(胃肠道和妇科肿瘤诊断的平均年龄约为 40-50 岁)。然而,报道了几例严重的病例,具有特征性结构性错配修复缺陷样特征,肿瘤发生于儿童期或青春期。聚合酶校对缺陷导致DNA复制错误的积累,引起体细胞高突变或超高突变(分别每兆碱基>10或>100个突变)和相关肿瘤特异性突变谱(突变特征)。
常染色体隐性遗传性腺瘤性息肉病综合征
碱基切除修复和错配修复基因双等位基因致病性变异导致隐性遗传性腺瘤性息肉病。因此,通过体细胞分析来识别错误的DNA修复突变特征,可以为此类肿瘤的原因提供线索。
MUTYH相关息肉病(MAP),发病率约为1/20000-60000,由碱基切除修复糖基化酶MUTYH双等位基因致病性变异引起。MUTYH相关息肉病与人群中常见的创始人突变有关,因此经常发生在血缘关系之外。杂合子的人群发生率约为1/50(1-2%),考虑到 MUTYH 杂合子携带者在人群筛查年龄发生结直肠癌的风险低至中度增加,在多基因panel检测中偶然发现的 MUTYH 杂合致病性变异不应被过度解释为致病性。在临床上,MUTYH 相关息肉病虽然是隐性遗传的,但与家族性腺瘤性息肉病的许多临床特征相似,典型的表型包括少于 200 个结直肠腺瘤。MUTYH相关性息肉病相关结肠外特征主要局限于十二指肠腺瘤(表 1)。
NTHL1 和 MBD4 编码碱基切除修复糖基化酶,其致病性变异可引起极其罕见的隐性息肉病。与MUTYH相关息肉病相比,其导致的综合征与一系列结肠外肿瘤相关(表 1)。NTHL1 肿瘤综合征的特征是胃肠道肿瘤、子宫内膜癌和乳腺癌的风险高,以及其他癌症类型和非肿瘤表现的风险增加。MBD4 相关肿瘤综合征导致急性髓系白血病(前期为骨髓增生异常综合征)、腺瘤性息肉病、结直肠癌以及葡萄膜黑色素瘤和神经鞘瘤(较少见)的风险增加。
结构性错配修复缺陷综合征(CMMRD)是由错配修复基因双等位基因致病性变异引起的,通常出现在儿童期。结构性错配修复缺陷综合征恶性肿瘤不局限于常见的林奇综合征相关癌症(表 1)。儿童中最常见的表现是血液系统恶性肿瘤和多形性胶质母细胞瘤。由于该综合征的严重性,为携带者提供了高强度监测方案。在患者中发现的大多数变异涉及较低外显率错配修复基因,例如 PMS2 和 MSH6。结构性错配修复缺陷综合征的诊断标准包括评估非肿瘤组织的错配修复功能,因此,原本在林奇综合征背景下被归类为意义不明的变异可能在结构性错配修复缺陷综合征背景下被重新归类为可能致病性。结构性错配修复缺陷样表型是由共存的错配修复基因和 POLE 或 POLD1 核酸外切酶结构域致病性变异引起的。
大规模外显子组测序研究在成人期发病的遗传性腺瘤性息肉病中发现了非林奇综合征错配修复基因(包括 MSH3 和 MLH3)双等位基因变异。这些基因单等位基因变异不会导致林奇综合征。
非腺瘤性息肉病表型
其他遗传息肉病综合征的表型之间存在大量重叠,而一些综合征主要表现为非腺瘤性肿瘤,包括锯齿状、错构瘤性和混合性息肉病。
锯齿状息肉病综合征是一种异质性疾病,其特征是一系列结直肠锯齿状肿瘤。过去认为锯齿状息肉病综合征是罕见的,但可能未被充分识别,筛查人群的发病率约为1/127-238。锯齿状息肉病综合征患者的一级亲属患结直肠癌的风险增加,但是总体不遵循经典的遗传模式。抑癌基因 RNF43 致病性变异是锯齿状息肉病综合征的罕见常染色体显性原因,解释了不到 3% 的锯齿状息肉病综合征病例。RNF43 是 Wnt 信号传导的负调节因子,RNF43 体细胞突变在通过锯齿状途径发生的散发性锯齿状息肉、腺瘤和结直肠癌中被发现。尽管一些研究表明,锯齿状息肉病综合征患者的多基因检测诊断率相对较高或中等,但许多变异可能是旁观者突变或代表另一种综合征的非典型表现。一些指南建议对锯齿状息肉病综合征患者进行基因检测,尽管主要是为了排除其他遗传疾病,因此,重点关注年轻患者、锯齿状息肉内有异型增生的患者以及有垂直传播证据的个体。
多发性错构瘤综合征包括幼年性息肉病综合征、Peutz-Jeghers综合征和PTEN错构瘤综合征,其具有不同的临床表型,但均为显性遗传(表 1)。关于幼年性息肉病综合征,SMAD4 和 ENG 致病性变异与遗传性出血性毛细血管扩张症和上消化道表型存在显著的基因型-表型相关性,而 BMPR1A 变异主要引起下消化道表型。一种称为婴儿期幼年性息肉病的严重形式是由 BMPR1A 和 PTEN 基因所在的染色体新发微缺失引起的。胃肠道错构瘤与延伸到息肉边缘的肌肉链的树状化有关,通常是有蒂的,在息肉切除术期间需要延长烧灼。Peutz-Jeghers 综合征由 STK11 胚系致病性变异引起,其特征是胃肠道多发错构瘤性息肉,患各种癌症的风险增加。然而,PeutzJeghers 综合征的表现主要与非肿瘤性并发症(与幼年性息肉病不同),特别是息肉诱发的肠套叠的风险有关,因此与儿童期肠切除的高概率相关。Peutz-Jeghers 综合征患者通常在一生中的早期出现皮肤黏膜色素沉着,在成年期消退,这一特征在临床上与其他皮肤黏膜色素沉着疾病(例如 Laugier-Hunziker 综合征)区分开来,Laugier-Hunziker 综合征发生于成年期,没有胃肠道表型。PTEN 错构瘤综合征主要是一种非胃肠道表型,由 PTEN 胚系致病性变异引起,与乳腺癌、甲状腺癌、子宫内膜癌和肾癌等肿瘤类型的风险增加有关,但在胃肠道中通常是非肿瘤性的。PTEN致病性变异导致不同的临床表型,包括临床定义的疾病,如Cowden综合征、Bannayan-Riley-Ruvalcaba综合征和Proteus-like综合征。
遗传性混合性息肉病综合征是一种多发性腺瘤、炎性息肉和错构瘤综合征,最初在一个德系犹太人大家族中被临床诊断出来。识别的遗传原因包括跨越 SCG5 基因 3ʹ 末端和 GREM1 位点上游区域的重复,导致 BMP 拮抗剂 GREM1 表达增加。在非德系混合息肉病患者或家族中报道了额外的 GREM1 5ʹ 重复。
此外,神经纤维瘤病等疾病患者可能出现神经源性息肉样病变,包括节细胞神经瘤,但这些在肠道没有已知的恶性潜力,尽管其可能导致旺炽性息肉病,给结肠镜监测潜在的肿瘤转化带来了挑战。
遗传原因不明的息肉病
多发性腺瘤表型
多发性结直肠腺瘤表型也称为不明原因结肠息肉病,定义为10-99个结直肠腺瘤(图 2)。多发性结直肠腺瘤表型在筛查年龄较大的人群(即 >60 岁)中相对常见,在年轻个体中很少遇到,反映了环境风险是重要因素。
队列研究评估了多发性结直肠腺瘤表型患者检测的诊断率。一项研究分析了遗传学实验室数据,调查了这种表型的患者是否具有 APC 和 MUTYH 双等位基因突变,结果显示,有 10 -19 个腺瘤的患者检出率为 9%,有 20-99 个腺瘤的患者为 17.2%。在一项对 3789 名患者多基因panel检测数据(≥14 个遗传性结直肠癌和息肉病基因)的分析中,7.6% 的 10-19 个腺瘤患者和 13.7% 的 20-99 个腺瘤患者检测到突变。病例系列研究显示,林奇综合征是多发性腺瘤患者中最常诊断的遗传特征。这些研究具有固有的转诊偏倚,因为包括转诊进行基因检测的患者。然而,腺瘤多发性的诊断率与结直肠癌个人或家族史之间似乎存在关联。
及时识别致病性变异有助于指导患者预防结直肠癌的决策,包括监测、内镜检查和手术,并提示有遗传致病突变风险的相关个体进行级联检测。
识别新的致病基因
几十年来,特别是大规模测序技术问世以来,致力于探索未解决的胃肠道息肉病的遗传原因。与非息肉病性结直肠癌不同,在发现新的息肉病易感基因方面取得了显著成功,鉴定了多个基因和综合征,其中许多在过去十年中发现(POLE、POLD1、NTHL1、MBD4、RNF43、MSH3、MLH3 和 GREM1)。然而,这些基因解释了个别息肉病病例,意味着许多息肉病病例,特别是锯齿状和增生性、腺瘤性或混合性息肉病,未识别遗传原因。在过去的十年中,提出了息肉病易感性候选基因,包括BMPR2、ERCC6、WRN、SMAD9、FOCAD、POLQ、MCM9、MCM8、WNK2、DSC2、PIEZO1、ZSWIM7和多个Wnt信号通路基因。然而,迄今为止收集到的关于这些基因的证据不足,在某些情况下相互矛盾,因此很难将其纳入临床分子诊断。
非遗传性息肉病危险因素
生活方式和环境暴露可促使结直肠癌的发生,在某些情况下,导致胃肠道息肉。吸烟与结肠息肉风险密切相关,与远端锯齿状息肉有关。然而,环境危险因素与腺瘤多发性的相关性尚未得到系统评估。不同组织学的治疗相关息肉病在息肉病患者中已有报道,这些患者胚系基因检测未发现相关变异,既往接受过腹部盆腔放疗或烷化剂化疗,用于治疗儿童或年轻成人癌症。由于其获得性(非遗传性)性质。治疗相关息肉病的识别也与患者亲属有关。
息肉病综合征肿瘤分子特征
多个息肉病致病基因是DNA修复机制的组成部分。由遗传性(胚系)或获得性(体细胞)突变引起,DNA修复缺陷在肿瘤中留下DNA损伤类型的特定突变指纹特征。配对肿瘤和对照组织显示错配修复缺陷(通过免疫组织化学或微卫星不稳定性检测)用于在患者中识别潜在的林奇综合征或结构性错配修复缺陷综合征。在结构性错配修复缺陷综合征中,非肿瘤组织错配修复功能检测是重要的辅助检测,有助于解释基因检测的不确定结果。
患者肿瘤NGS检测(基因组测序、外显子组测序或多基因panel检测)可用于评估肿瘤突变负荷和是否存在与DNA修复缺陷相关的特定突变特征。这些DNA修复缺陷相关特征(表 2)可能有助于解释临床意义未明的变异,以及识别该综合征表型谱的肿瘤。DNA修复缺陷引起的高肿瘤突变负荷是对免疫治疗反应良好的预测指标。
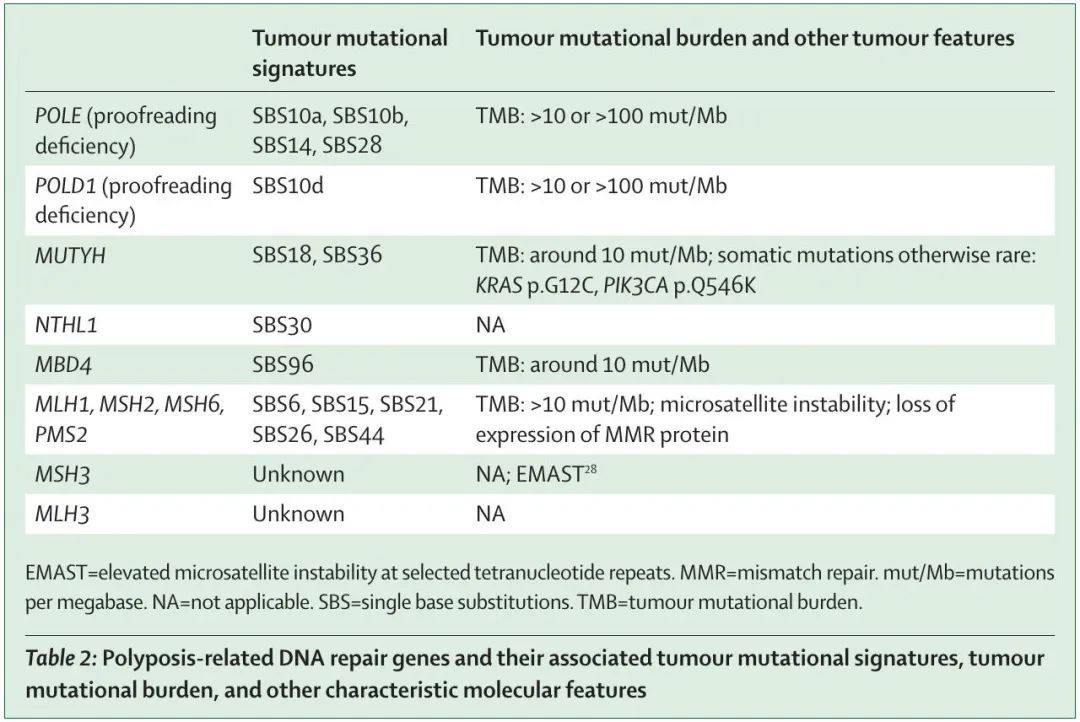
表2
对于隐性综合征,甚至是常染色体显性遗传性癌症综合征,如果使用灵敏度高的测序技术,这些突变标志可以在非肿瘤组织(包括血液DNA)中检测到。
基因检测
目前大多数分子诊断方法依赖于市售或定制多基因panel检测,包括大约 80-200 个癌症易感基因。在一些卫生系统中,使用了更多表型特异性基因panel。当多发性腺瘤表型定义为 ≥10 个累积腺瘤时,息肉病患者多基因panel检测的诊断率约为 1/9,腺瘤多发性增加、个人或家族癌症史以及年龄较小与多基因panel检测的诊断率增加相关。
在实践中,鉴于大多数息肉病相关基因包含在常规基因检测使用的多基因panel中,以及基因型-表型相关性的不可预测性,建议对所有息肉病和结直肠癌相关基因进行全面检测,该方法可克服罕见息肉类型误诊或组织学分型不完整的挑战。
一些超罕见和最近发现的基因,例如 MBD4、NTHL1、MLH3、MSH3、AXIN2、RNF43 或 ENG,可能不常规包含在临床可及的多基因panel检测中。考虑到相关综合征的预期发生率极低,对诊断性能的影响应该很低。然而,当患者的表型、肿瘤分子特征或两者都提示或支持特定的遗传原因时,我们建议对相关基因进行单独分析(如果该基因未包含在panel中)。例如,息肉病与急性髓系白血病、葡萄膜黑色素瘤、神经鞘瘤或这些疾病的组合共存的患者进行MBD4 检测,诊断为少牙畸形的患者进行AXIN2检测,遗传性出血性毛细血管扩张症和幼年性息肉患者在没有 SMAD4 致病性变异的情况下进行ENG检测,或在存在提示特定DNA修复缺陷的肿瘤突变特征或分子特征的情况下检测相应的 DNA 修复或聚合酶基因。
对启动子和内含子区域的分析可能揭示影响基因调控元件的突变和非典型剪接变异。然而,这种方法的相关性和成本效益仍未被探索,有时甚至有争议。DNA和RNA双组学多基因panel检测已被证明可以改善遗传性癌症基因的诊断结果,辅助解释推定的剪接变异,识别非典型剪接变异,发现等位基因特异性表达事件(等位基因表达缺失),提示调控区域或启动子甲基化。
体细胞嵌合体是息肉病中相对常见的事件,尤其影响APC,在错构瘤性息肉病基因(PTEN,STK11,BMPR1A和SMAD4)中也有报道。当变异在胚胎组织中新发时发生嵌合。取决于突变在胚胎发育的多早期发生,以及结肠上皮和其他靶组织嵌合的程度,相应的表型可能较温和或与典型的结构性病例相似。嵌合体的识别具有挑战性,在分析多基因panel测序数据或检测其他组织(例如正常结直肠粘膜)时,需要降低变异检测阈值(至变异丰度约5%),因为嵌合变异可能不存在于造血细胞中。在息肉病的情况下,对同一患者多个息肉检测此类变异将识别疾病相关结肠组织(很容易从息肉病患者的结直肠监测中获得)中的低水平嵌合体。
一些致病性变异的提示或解释是微妙的:对于 POLE 和 POLD1,只有影响蛋白质核酸外切酶结构域的变异才应考虑为潜在致病性,功能缺失(截短)变异或位于核酸外切酶结构域之外的任何类型的变异与息肉病和癌症综合征无关。应考虑 APC 启动子 1B 分析,尤其是胃近端息肉病(胃底腺)和胃腺癌患者。与混合性息肉病相关的 GREM1 改变包括 GREM1 上游大型重复,导致基因表达增加。导致林奇综合征的 EPCAM 改变是位于 EPCAM 下游的大型 3ʹ 基因缺失,通过使 MSH2 启动子甲基化导致基因沉默。
对于所有基因,应按照美国医学遗传学和基因组学学会和分子病理学协会变异分类指南进行变异分类。如果可及,应应用 ClinGen 变异管理专家小组制定的基因特异性建议。
临床管理
癌症预防和早期发现
胃肠道监测和手术
对息肉病患者进行结肠镜监测可以预防结直肠癌及降低结直肠癌相关死亡率。全球息肉病数据库研究显示,在未参加基于数据库的结直肠筛查计划的患者中,结直肠癌发生率为50-70%,在参加基于数据库的监测的患者中,为3-10%。息肉病患者或专业临床部门数据库与高质量的管理有关,包括内镜和手术管理,对高危个体具有有效的召回机制。一些专业中心还提供多学科整体终身管理,提供最佳决策。
鉴于家族性腺瘤性息肉病是一系列在临床表型上具有很大差异的疾病,部分但不完全由基因型解释,预防性结直肠手术的时间、年龄和范围可能与临床表现有关。家族性腺瘤性息肉病的偶然诊断通常与较轻的表型有关,可能在患者因不相关的医学问题进行多基因检测时发现。这些患者的自然病程尚未阐明,可能不一定需要立即进行结肠切除术。
因此,建议采用个体化方法来监测家族性腺瘤性息肉病和其他息肉病综合征。尽管大多数共识指南建议,对于决定不立即手术的家族性腺瘤性息肉病,每 1-3 年进行一次结肠镜检查,但这些间隔和转为手术切除的决定取决于腺瘤负荷的逐步变化。结肠镜监测能够评估腺瘤负荷和分布,从而指导所需的预防性手术的时间和类型。染色内镜检查可用于息肉病综合征诊断时确定表型。然而,不建议在长期监测中使用染色内镜,因为可能导致微腺瘤的过度诊断,而微腺瘤与手术决策无关。在 MUTYH 相关息肉病的情况下,监测显示一段时间后癌高风险可能表明癌变加速,提示尽管与大多数家族性腺瘤性息肉病患者相比,腺瘤数量较低,结直肠癌终生外显率较低,但仍可能需要较频繁的监测。
关于手术,选择通常为全结肠切除术加回肠直肠吻合术或直肠结肠切除术和回肠储袋肛管吻合术。取决于直肠息肉的大小和数量、是否存在高度异型增生、基因型以及手术的功能结果。与接受更广泛手术的患者相比,接受结肠次全切除术的患者通常具有更好的肠功能,但必须仔细评估风险,结合疾病特异性和患者特异性因素。结肠切除术后有残留结直肠或回肠肛管储袋的患者需要终生监测,因为存在异时性癌症的风险。
很少进展为癌症的十二指肠息肉,和上消化道息肉发生于家族性腺瘤性息肉病和其他息肉病综合征(表 1)。根据共识指南,可能需要进行特定的上消化道监测。在家族性腺瘤性息肉病中,十二指肠息肉病的终生外显率接近 100%,但只有 5% 会发展为十二指肠癌。因此,进行上消化道监测,只有当这不再可行时,患者通常才会接受预防性十二指肠切除术,通常通过结合分期系统(如 Spigelman 分类)来确定。内镜识别胃腺瘤病变具有挑战性,胃腺瘤和壶腹肿瘤的进展可能独立于 Spigelman 分类。
小肠肿瘤可发生于家族性腺瘤性息肉病,是一项临床挑战。目前的美国和欧洲指南不建议对家族性腺瘤性息肉病进行十二指肠以外的小肠监测。很少有数据支持在回肠储袋肛管吻合术监测之外或在 Whipple 手术前的背景下,对 Treitz 韧带以外的小肠进行监测。
由于人群的异质性,未发现致病基因的多发性结直肠腺瘤患者的结直肠癌风险管理仍然具有挑战性。考虑到风险因素可能包括多因素和未识别的遗传综合征,建议采用个体化方法。由于一部分多发性结直肠腺瘤患者(约 10-20%)具有上消化道表型,通常伴有十二指肠腺瘤,上消化道评估是合适的。对于多发性结直肠腺瘤患者的一级亲属,临床医生可能考虑在转诊时进行结肠镜风险评估,以评估家族表型,然后再确定进一步监测。
表 3 总结了美国、英国和欧洲息肉病综合征上和下消化道内镜监测指南。
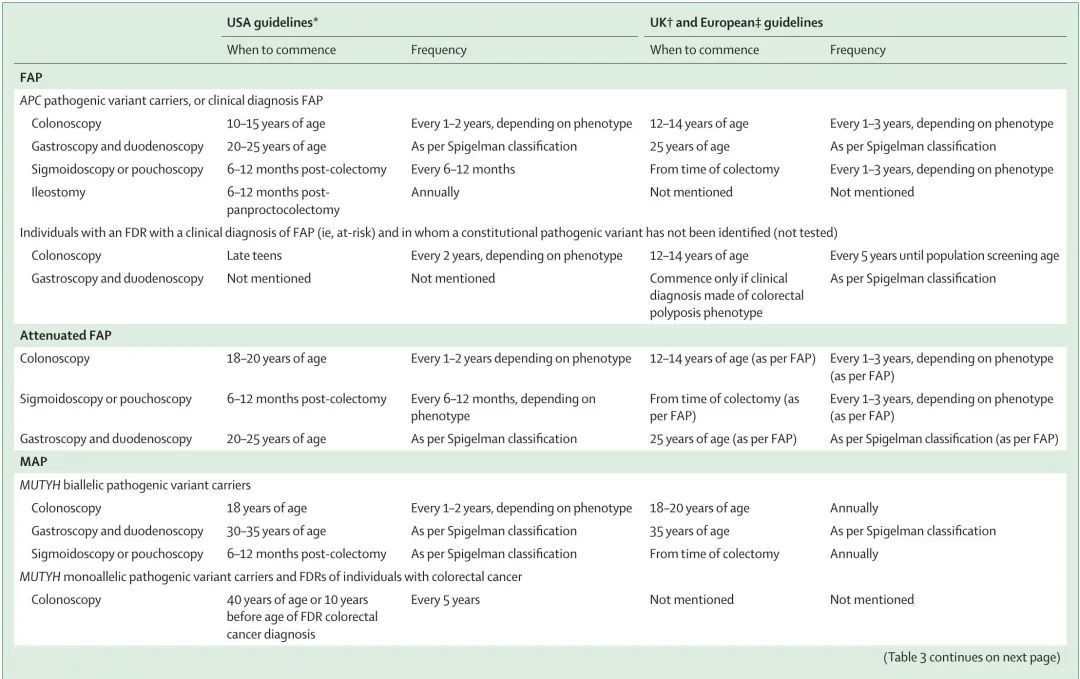
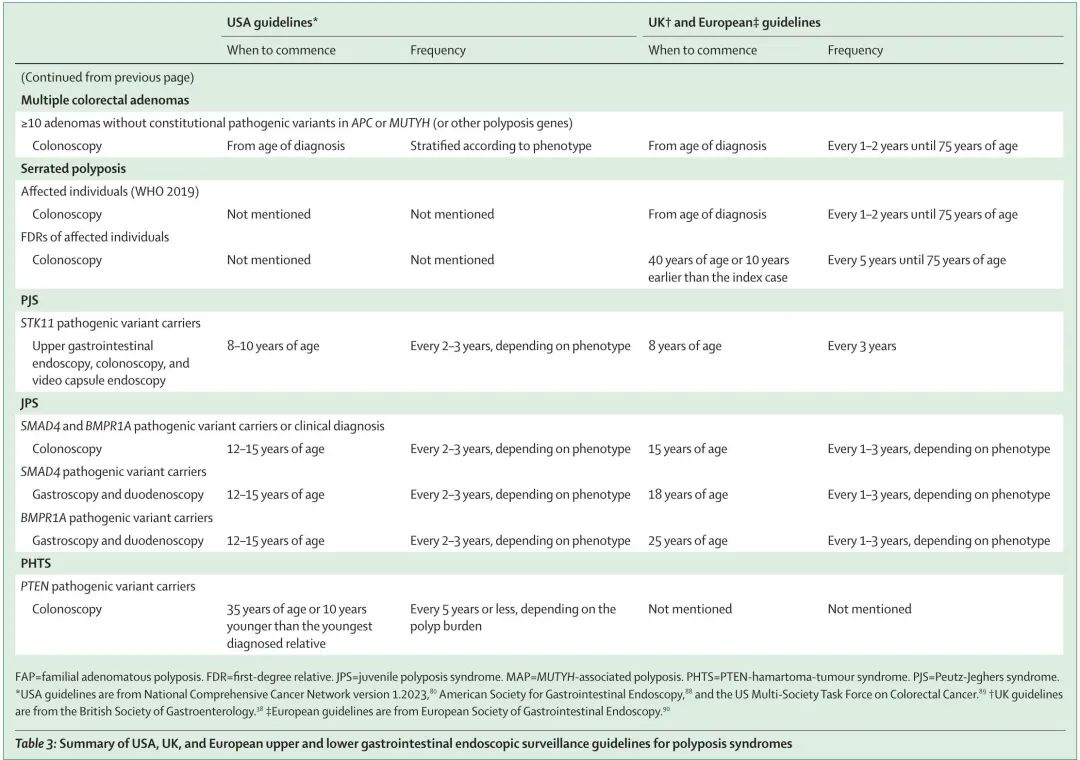
表3
胃肠道外癌症监测
胃肠道外癌症监测应遵循特定国家/地区的建议(如果可及),尽管关于这种监测有效性的证据较少。对于极罕见或最近发现的息肉病综合征,尚无共识建议,应根据已发表的证据进行胃肠道外癌症监测。
药物预防
降低癌症风险是息肉病综合征药物预防的最重要目标。在家族性腺瘤性息肉病中,已经使用了各种替代终点,包括息肉多发性。然而,关于家族性腺瘤性息肉病的这些终点是否具有临床意义,仍存在一些争议。腺瘤可能代表接受内镜监测的患者癌症风险的生物标志物,尽管大多数腺瘤不会进展为癌症,因此腺瘤多发性的减少可能不是疾病改变的有力证据。然而,一些令人信服的研究表明了这些替代终点的改变,主要使用了非甾体抗炎药。在一项随机对照试验中,主要终点为大型手术、晚期肿瘤或十二指肠疾病进展的综合,使用依氟鸟氨酸加舒林酸与疾病进展延迟有关。
疫苗
已经开发了移码肽新抗原疫苗,可诱导肿瘤抗原特异性免疫反应,以预防错配修复缺陷肿瘤,例如林奇综合征或CMMRD相关肿瘤,并已被纳入临床试验。一项小鼠模型临床前研究表明,肽疫苗接种联合非甾体抗炎药(如阿司匹林或萘普生)可能减轻肿瘤负荷和延长生存期。
个体化治疗
免疫检查点抑制剂可有效治疗错配修复缺陷的结直肠癌和其他肿瘤类型,因此可用于治疗林奇综合征和CMMRD相关癌症。与错配修复缺陷癌症一样,聚合酶ε和δ校对缺陷的肿瘤,无论错配修复功能完整还是缺陷,都为高突变,产生高肿瘤新抗原负荷,导致强大的免疫浸润。这些肿瘤的高免疫原性表型转化为较好的预后和对免疫检查点阻断的反应。在 MUTYH 缺陷和 MBD4 缺陷癌症中观察到类似的结果,免疫检查点抑制剂已显示出有希望的治疗潜力。预计 NTHL1 缺陷肿瘤也有类似的反应,尽管缺乏已发表的研究。
长期以来,我们对罕见遗传性结直肠癌综合征(如家族性腺瘤性息肉病)的了解为APC等基因在散发性结直肠癌中的作用以及肿瘤治疗的潜力提供了见解,这超出了遗传性综合征本身。使用氨基糖苷类或奈加霉素等药物可以实现 APC 中提前终止密码子的通读,从而恢复癌细胞系中 APC 的生物活性。家族性腺瘤性息肉病小鼠模型中大环内酯类药物ZKN-0013(一种通读剂)治疗导致肠腺瘤显著缩小,对于由APC基因无义突变引起的家族性腺瘤性息肉病显示出有希望的治疗潜力。
porcupine抑制剂可能消除 Wnt 通路基因(包括 RNF43)异常表达,一项早期临床试验表明,这些抑制剂联合靶向免疫疗法对 RNF43 突变的结直肠癌患者有效。
mTOR抑制剂在PTEN突变或STK11突变癌症的治疗中显示出有希望的结果。特别是,mTOR抑制剂用于PTEN错构瘤综合征成人患者,在整个单臂试验期间显示出多系统标志和症状的改善。目前正在进行mTOR抑制剂治疗PTEN错构瘤综合征的临床试验(NCT02991807和NCT04094675)。此外,mTOR抑制剂治疗导致PTEN-BMPR1A缺失的婴儿期幼年性息肉病患者的肠病、肠道出血和结肠切除率降低。
总 结
在过去的一个世纪里,对导致胃肠道腺瘤多样性的一系列遗传综合征的认识不断扩大。随着基因发现转化为临床实践,可以通过准确的风险评估、个体化的内镜和预防性手术管理以及有效的亲属级联检测,提供更精确的基因特异性癌症预防策略。此外,改进的分子诊断和有效的肿瘤学管理,结合其他患者因素,有助于决策,改善患者预后。对特定基因型-表型关系、与这些综合征相关的体细胞事件以及特定遗传风险因素的外显率的了解,以及对分子诊断者中存在的临床异质性的了解,推动了这些进展。有效的临床管理需要专业知识和多学科管理。尽管识别了罕见的息肉病病因,但其使用多基因panel检测在人群中很少检测到,随着临床实践中全基因组测序的出现,有可能识别家族性胚系变异。在这些情况下,对体细胞分子机制(如DNA碱基切除修复)的了解,可能指导临床管理,包括监测和癌症治疗。
尽管近年来取得了进展,但仍然存在巨大的知识空缺:识别其他可能非常罕见的高外显率息肉病遗传综合征;探索多基因风险和遗传修饰;了解息肉病病因中遗传与环境和生活方式相关风险因素之间的相互作用;以及应对诊断挑战的方法,例如应该如何实施和标准化体细胞检测,以指导息肉病患者的分子诊断和临床管理,包括识别嵌合病例。了解如何区分由遗传风险或环境和生活方式相关风险因素引起的病例,可能会在患者的个体化管理方面提供临床益处。内镜监测vs手术治疗的时间、频率和相对益处仍主要基于专家意见或小队列研究的观察数据。需要使用替代终点来评估药物预防在癌症预防中的益处,这是一项挑战。这些和其他未解决的问题将指导该领域未来的研究,定义临床实践的未来变化。
参考文献:
Valle L, Monahan KJ. Genetic predisposition to gastrointestinal polyposis: syndromes, tumour features, genetic testing, and clinical management. Lancet Gastroenterol Hepatol. 2024 Jan;9(1):68-82. doi: 10.1016/S2468-1253(23)00240-6. Epub 2023 Nov 4. PMID: 37931640.

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#基因检测# #胃肠道息肉病#
30