
复旦大学领衔Nature:首篇!36个中国民族种群116个个体的泛基因组,发现隐藏的特殊基因序列
2023-06-23 BioMed科技 BioMed科技 发表于上海

通过联合努力,研究团队成功测序了36个中国少数民族和汉族样本,并构建了116个高质量的基因组组装。
在过去的二十年里,参考人类基因组序列一直作为遗传和生物医学研究及应用的基础,但广泛共识认为没有单一的参考序列能够代表全球人群的基因组多样性。
根据研究数据显示,仅有47个样本构建的人类全基因组参考(HPRC)在东亚人群样本的代表性较低,其中仅有3个南方汉族样本被包含在HPRC参考中,对于中国这样一个拥有14.4亿人口、众多民族群体的地区来说,代表性不足。此前的研究还表明,亚洲的遗传多样性在1000人基因组计划等大型国际合作项目中得到的覆盖较少。
基于此,26家科研机构包括复旦大学、西安交通大学和中国医学科学院等,共同组成了中国人群泛基因组联盟(CPC)。通过联合努力,研究团队成功测序了36个中国少数民族和汉族样本,并构建了116个高质量的基因组组装。
该成果以"A pangenome reference of 36 Chinese populations"为题,于2023年6月14日发表在Nature杂志上,第一通讯作者为中国科学院特聘研究员徐书华。

【多样性中国基因组的组装】
研究团队介绍了中国全基因组联合研究计划的第一阶段数据,其中包括116个高质量和单倍型相位化的全新组装序列,基于代表36个中国少数民族群体的58个核心样本。
这些中国全基因组联合研究计划的核心组装序列具有平均30.65倍高保真长读长测序覆盖度,平均连续性N50超过35.63兆碱基,并且平均总大小为3.01吉字节。这些核心组装序列为GRCh38参考基因组添加了1.89亿碱基对的有色体多态序列和1,367个蛋白编码基因复制。
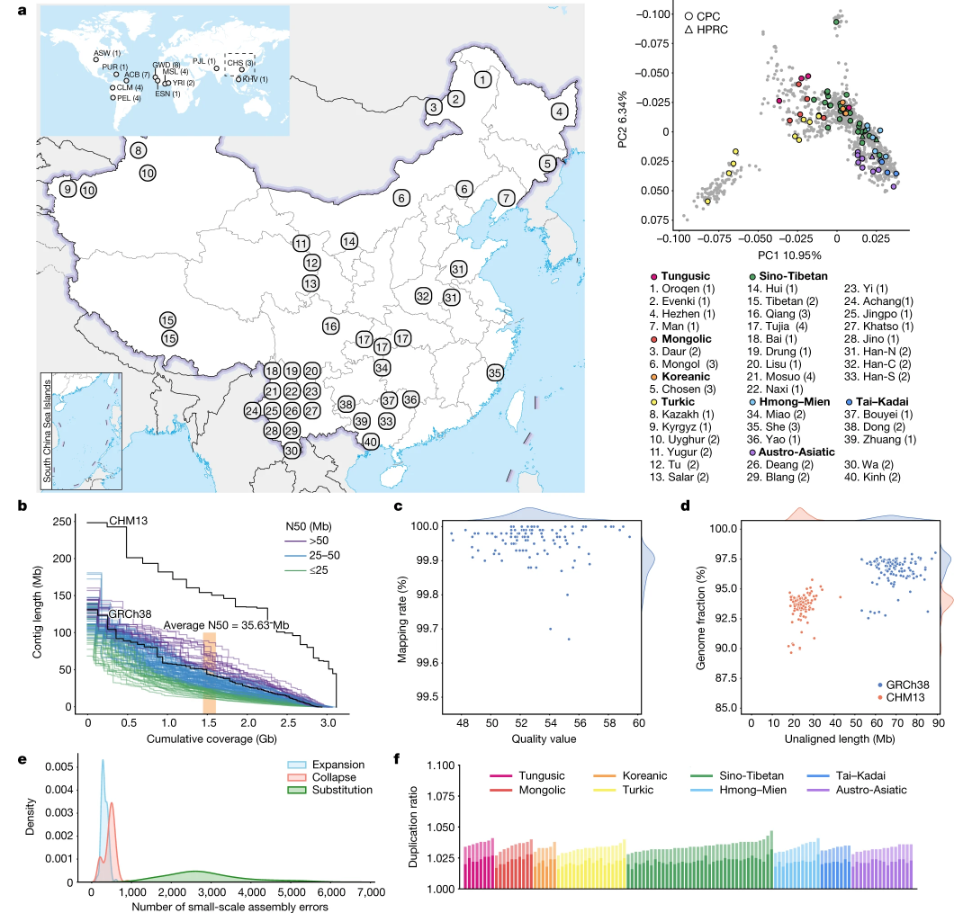
图1 CPC小组使用58个核心样本的二倍体组装得到了CPC面板
【CPC组装序列的基因组特征】
研究团队使用Phased Assembly Variant caller对116个组装体与T2T-CHM13进行比对,并对每个样本进行变异调用。根据插入频率,确定了115个基因组区域作为插入热点,总长度为204.8Mb。
然后,研究团队对每个组装体相对于GRCh38参考基因组进行了拷贝数变异(CNVs)的注释。在所有组装体中,共有1,367个蛋白编码基因在至少一个基因组中拷贝数增加。每个组装体平均观察到53个(27-100个)具有拷贝数增加的基因,其中13.39%的CNV基因在整个CPC组装体集中的频率超过5%,而57.86%的CNV基因仅在单个单倍体中发现。
研究团队发现CPC组装体集中有1,079个重复基因,在HPRC组装体中没有观察到。在CPC组装体集中发现的562个(52.1%)新重复基因根据全基因组关联研究(GWAS)目录与特征相关联,其中207个(19.2%)与至少一种由疾病本体注释的人类疾病相关联,这表明CPC组装体集在疾病和表型关联研究中具有显著的潜力。
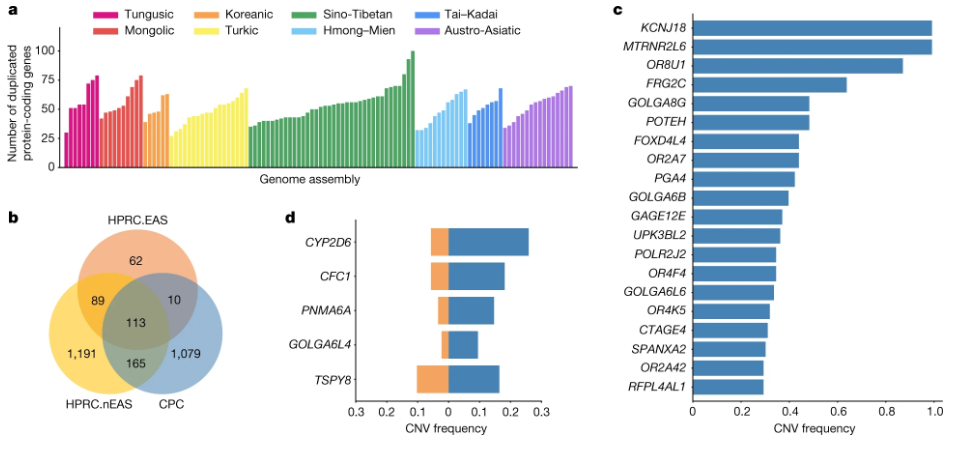
图2从CPC组装中识别出了拷贝数变异
【CPC全基因组的变异】
研究团队使用Minigraph-Cactus流程构建了CPC泛基因组的变异图谱,其中单倍型组装体可以表示为由序列节点组成的不同路径。
此外,他们发现在所有单倍型中,有4.96Mb的非参考序列出现在≥95%的单倍型中,代表了样本群体的核心基因组,还有72.24Mb的非参考序列出现在≥5%且<95%的单倍型中,代表了共有的基因组。
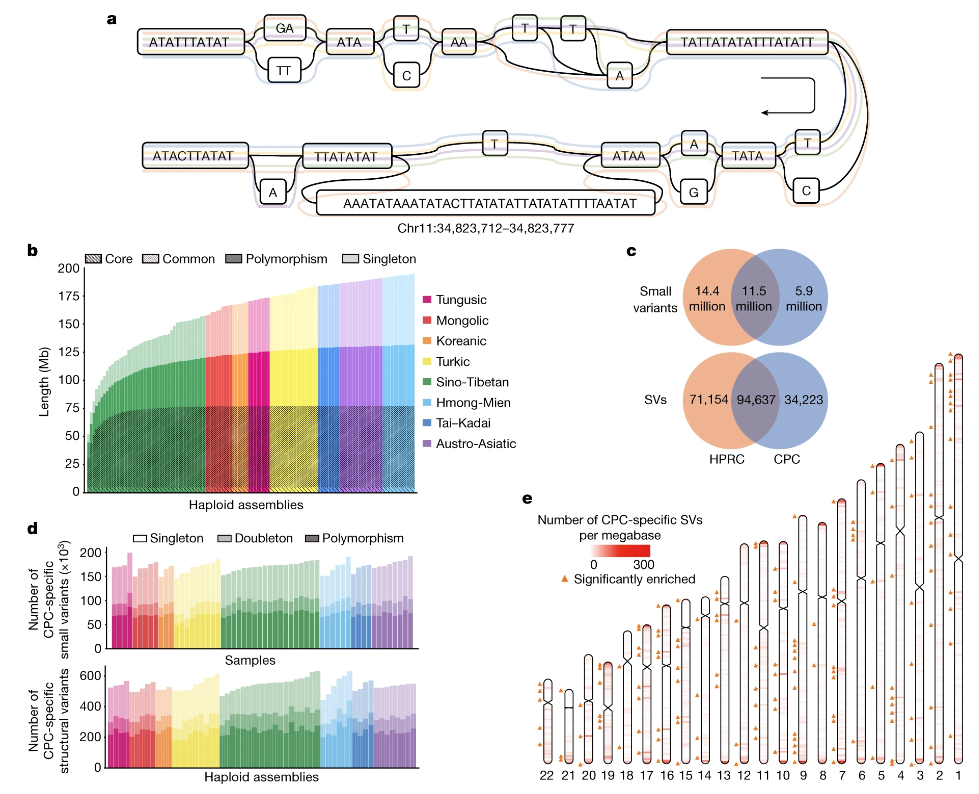
图3 将CPC 泛基因组图与HPRC组装进行比较,分析了CPC特有变异和CPC特定变异
【使用CPC参考进行短读映射】
为了进行变异调用,研究团队将图谱参考坐标中的GAM文件与线性参考坐标中的BAM文件进行了映射。结果显示,所有样本的映射率平均下降了0.58%(0.54-0.61%)。于是推测,当使用传统的线性参考进行调用或记录变异时,图谱参考的优势会丧失,因为图谱参考中的新序列在线性坐标中是缺失的。
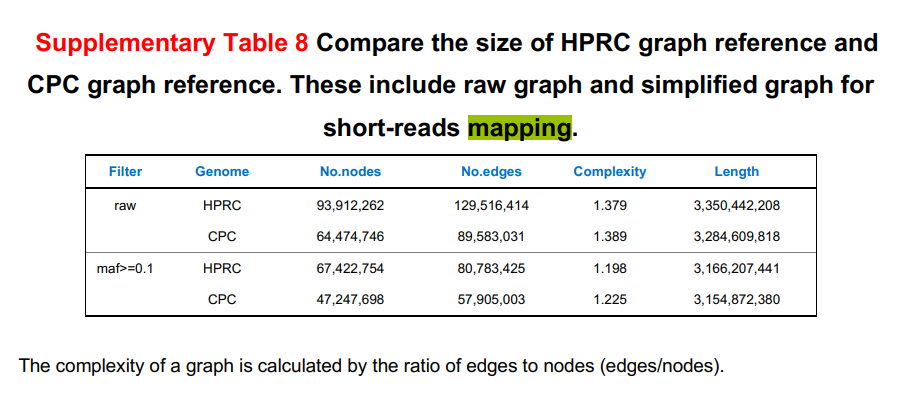
表1 比较了HPRC图谱参考和CPC图谱参考的大小,包括用于短读映射的原始图和简化图
【与人类全基因组参考联合研究计划(HPRC)全基因组图的比较】
研究团队还鉴定了1590万个小变异和78,072个结构变异,其中590万个小变异和34,223个结构变异在最近发布的全基因组参考序列中尚未报告。
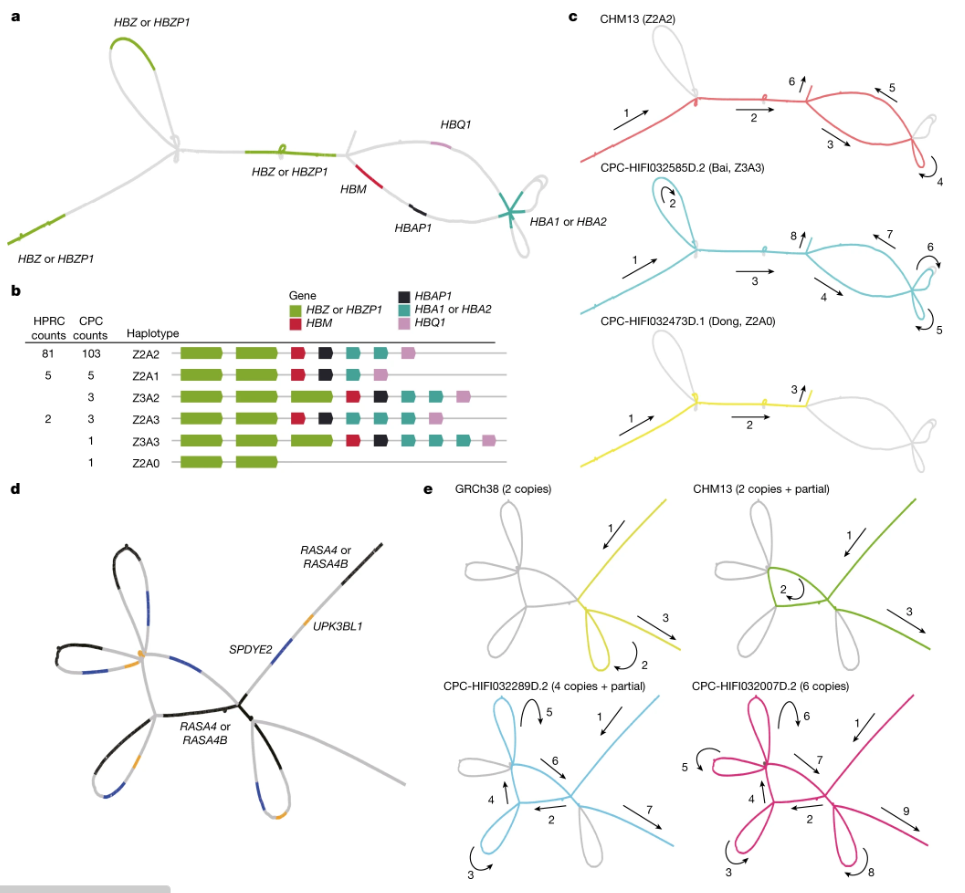
图4 在CPC 泛基因组图中可视化了新颖和复杂的结构变异。
【古代基因渗入和注释】
中国全基因组联合研究计划的数据显示,当从代表少数民族群体的人中获取数据时,可以显著增加对新颖序列和缺失序列的发现。这些缺失的参考序列富集了古代遗传位点的等位基因和与角化、对紫外线辐射的响应、DNA修复、免疫反应和寿命相关的基因,因此对于揭示人类进化以及在复杂疾病遗传性研究中找回缺失遗传性具有重要意义。
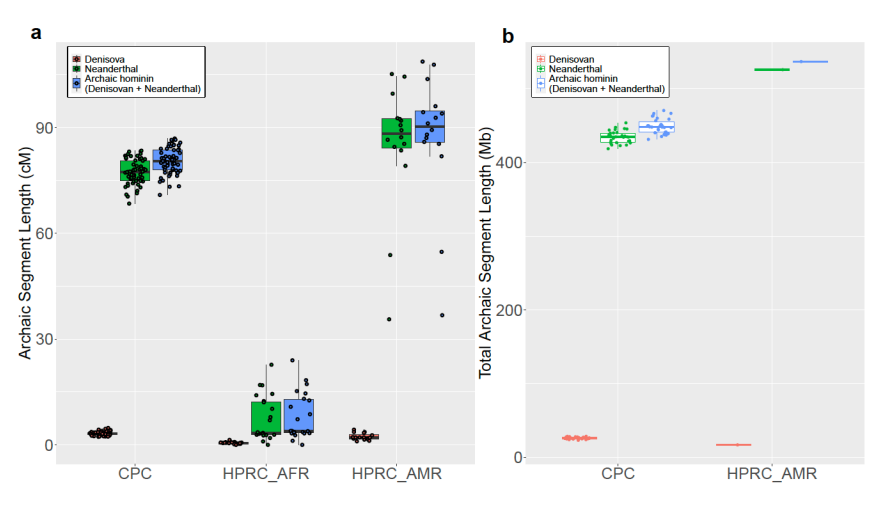
图5 比较了CPC中检测到的旧石器时代基因渗入片段的总长度与HPRC中的长度。

图6 比较了CPC研究中东亚人群的旧石器时代基因渗入片段的累计长度。
【研究意义】
徐书华的团队在基因组学领域取得了重要的突破,并通过前沿的技术和分析手段研究了亚洲人群的遗传演化历史。他们的工作不仅关注中国人群,还涵盖了周边国家和地区的人群。通过深入研究这些人群的遗传背景和基因组多样性形成机制,他们希望能够揭示人类族群的演化历史以及遗传变异对人类基因组序列功能和表型意义的影响。
这种群体基因组学的研究不仅帮助我们了解人类的起源和演化过程,还对现实的人类健康具有重要的参考意义。通过回溯族群的“基因史”,我们能更好地理解复杂的遗传疾病,并为精准医学的发展奠定群体遗传学基础。未来,随着大数据的积累和精准医学的发展,我们有望实现更加个性化和精确的医疗,根据不同人群的基因组差异来制定个性化的治疗方案。
参考文献:
1.https://doi.org/10.1101/2022.07.09.499321
2.https://www.science.org/doi/10.1126/science.1177074
3.https://doi.org/10.3389/fgene.2013.00127
原文链接:
https://www.nature.com/articles/s41586-023-06173-7
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





