
加州大学Teresa H. G.小组JCTC期刊论文:通过学习蛋白质内部坐标之间相关性来高质量重建3D笛卡尔坐标
2023-08-13 ComputArt计算有乐趣 ComputArt计算有乐趣 发表于浙江省

在该项工作中,研究人员开发了一种新的机器学习方法来提高3D 笛卡尔坐标反向变换的保真度。
生物大分子的结构通常使用两种数学表示法来描述:内部坐标和笛卡尔坐标。内部坐标表示由一组键长、键角、二面角或扭转角定义,笛卡尔坐标表示则由欧几里得空间中所有原子的x、y、z 坐标定义。两种表示法在不同的应用场景中均已被广泛应用。内部坐标是核磁共振结构测定和精修的首选描述,有利于几何优化,而笛卡尔坐标是分子动力学模拟和X 射线晶体学结构的首选格式。当所有键长及角度都被精确指定时,使用自然延伸参考系(NeRF)等算法,可以从内部坐标进行反向变换重建得到蛋白质骨架结构3D 笛卡尔坐标,在此过程中通常会将键长和角度固定在平均值以降低结构建模的复杂性。直觉表明这种固定键长与角度的方法能够很好地重建笛卡尔坐标,因为键长与角度的值都被约束在平均值附近,仅存在很小的误差。但事实上,有研究表明与平均值的微小偏差也会极大程度上影响笛卡尔重建,对于平均包含150个氨基酸的蛋白质,在固定键长和角度的条件下,内部坐标反向变换到主链Cα原子笛卡尔坐标时产生的 RMSD 误差为 ∼6 Å。目前已有多项研究报道采用以PDB中序列或结构相关性(例如拉氏图中φ 和 ψ 扭转角相关性、Dunbrack转子库中使用的主链和侧脸扭转角相关性等等)为条件的统计方法降低笛卡尔重建的误差,但这些研究中都没有考虑到内部坐标中ω扭转角依赖性。
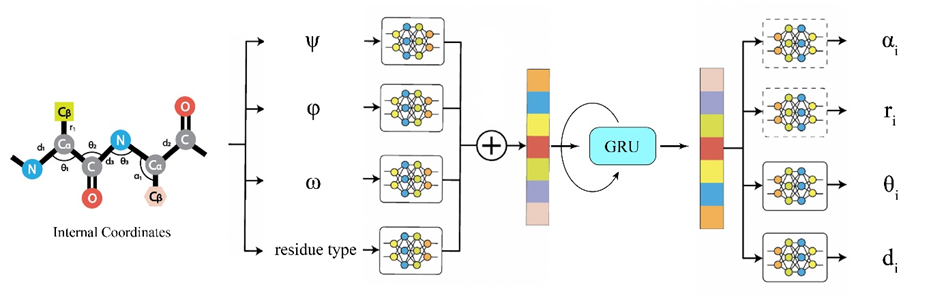
加州大学Teresa Head-Gordon小组与多伦多大学Julie D Forman-Kay小组针对这一问题,提出了一种更全面的机器学习方法——Int2Cart,可以量化地学习更深层次氨基酸序列上下文中的内部坐标相关性,相对于固定键长与角度标准假设,Int2Cart能够提供更准确的 3D 笛卡尔坐标。近日,该项研究工作发表在美国化学会出版的Journal of Chemical Theory and Computation期刊上 (Journal of Chemical Theory and Computation 2023 19 (14), 4689-4700)。【1】
研究人员提出的神经网络Int2Cart,架构如图1所示。该神经网络是门控循环单元(GRU)循环神经网络,其中每个时间步的输入是来自高斯修饰的φ、ψ 和ω 扭转角以及嵌入的残基类型共同构成的串联隐藏向量,GRU 的隐藏向量输出与不同的输出网络连接以分别预测主链键长d(例如N-Cα键长)、键角θ(例如C − N − Cα角),或可选地预测侧链键长r(例如Cα – Cβ键长)和侧链键角α(例如N − Cα – Cβ角)。
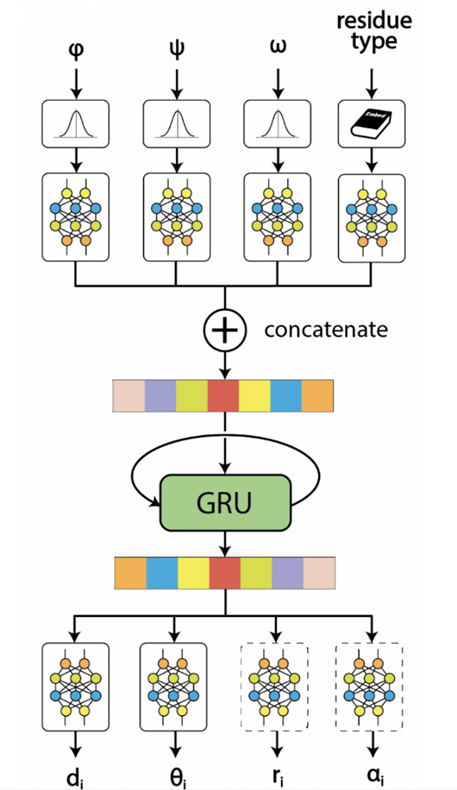
图1: Int2Cart模型架构
在中心组件GRU中,研究人员将GRU层堆叠三次,每层都包含一个隐藏状态ht,其信息由输入向量通过以下等式构成的重置和更新机制进行更新:
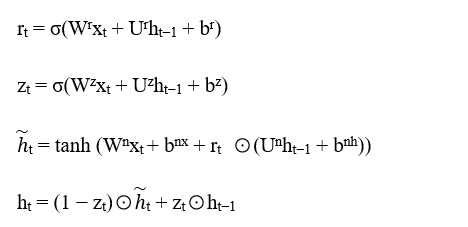
其中W, U, b是模型的可训练参数,xt是当前时间步的单元格输入,rt和zt表示重置和更新门,控制新的更新向量t中保留多少信息,并控制新隐藏状态向量ht由更新向量t和旧隐藏状态ht−1的组成方式。σ表示sigmoid函数,⊙表示逐元素乘法。
第一层GRU单元的输入为φ、ψ 和 ω 扭转角以及氨基酸类型。每个扭转角由高斯涂抹函数(Gaussian smearing function)离散为长度为180的向量,每个修饰后的扭转角向量进一步通过两个全连接层(维度为90和64)以及修正线性单元(ReLU)激活层进行变换,以生成扭转角的潜在表示。残基类型由可训练的嵌入字典进行编码,并形成长度为64的潜在向量而后通过两个全连接层(维度为128和64)以及ReLU激活层构成 GRU 单元的输入。最后一个 GRU 层的隐藏状态输出与后续多个输出连接,以预测主链键长和键角(或可选地预测侧链键长和键角)。每个输出都接入一个全连接神经网络,隐藏层维度大小为100,使用ReLU激活,输出层维度大小为1,无激活。
在获得主链扭转角以及模型预测的键长及键角数据后,研究人员使用SidechainNet 包重建蛋白质所有主链原子的3D笛卡尔坐标。SidechainNet利用自然延伸参考系(NeRF)算法,用前三个原子的位置以及新的键长、键角和扭转角按次序计算下一个原子的位置。
研究人员随后测试了在给定三个扭转角φ, ψ, ω的条件下,固定键长和键角与从Int2Cart学习键长键角两种方式重建笛卡尔坐标的误差情况。结果表明,与固定方法相比,Int2Cart重建的结构质量明显优于固定方法,结构RMSD分布以较低的RMSD值为中心,当所有蛋白质标准化为100个氨基酸时,中位RMSD为 2.14 Å,整个测试集的平均RMSD为3.5 Å。相比之下,当所有蛋白质标准化为100个氨基酸时,固定方法产生的中位RMSD为 3.24 Å,整个测试集的平均值为5.1Å。此外,Int2Cart表现还明显优于之前的研究,表明更深层次的序列相关性有利于模型学习。随后,研究人员还对Int2Cart与固定方法在整个测试集的回转半径(Rg)以及二级结构恢复率(SS-match)方面进行了进一步的分析。结果表明,尽管Int2Cart笛卡尔重建预测的结构的Rg值与真实结构更接近,固定方法笛卡尔重建方法仍然能够产生相对较好的结果。但需要注意的是,Int2Cart在二级结构恢复率方面显著地优于固定方法,Int2Cart预测结构的二级结构恢复率大于0.8的蛋白质所占比例有明显的提高。
研究人员还对模型在两个外部测试集上的表现进行了评估,评估模型的泛化能力。与之前的测试数据集结果相比,在CASP-12蛋白数据集上除了C-N-Cα角的预测结果在RMSE和相关系数方面变得稍差,所有其他预测目标都非常接近先前的结果。同时,在CASP12 测试集中标准化为 100 个氨基酸的蛋白质的重建结构RMSD 为 2.06 Å,与原始测试集的结果一致。
【总结】在该项工作中,研究人员开发了一种新的机器学习方法来提高3D 笛卡尔坐标反向变换的保真度。Int2Cart算法利用门控循环单元神经网络来预测给定扭转角的完整蛋白质序列中每个残基的键长和键角。结果表明,Int2Cart在测试集上重建的结构有95%在RMSD方面显著优于固定定主链键长和键角方法(固定主链键长和键角是多种蛋白质建模方法中的标准做法)。Int2Cart在CASP-12数据集上的成功进一步验证了Int2Cart 算法可以在不同类型的蛋白质之间转移,并且可以持续提高笛卡尔结构重建的质量。当前形式的模型提供了一种有用的计算工具,可以大大提高仅从主链扭转角重建的蛋白质结构的质量,无论是球状折叠蛋白质还是无序蛋白质。Int2Cart有望在蛋白质结构修正以及蛋白质力场的开发中得到广泛应用,这些蛋白质力场的开发会受益于模型预测的更准确的主链键长和键角。最后,Int2Cart GRU神经网络模型也可用于其他链分子(如核酸和脂质等),研究者只需使用新数据进行重新训练。
参考文献
【1】Li, J.; Zhang, O.; Lee, S.; Namini, A.; Liu, Z. H.; Teixeira, J. M. C.; Forman-Kay, J. D.; Head-Gordon, T. Learning Correlations between Internal Coordinates to Improve 3D Cartesian Coordinates for Proteins. J. Chem. Theory Comput. 2023, 19 (14), 4689–4700.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



