
华大基因开发新型空间染色质可及性测序工具SCA-seq,以单分子分辨率捕获表观遗传信息同时解析基因组构象
2023-12-22 测序中国 测序中国 发表于上海

SCA-seq利用纳米孔技术对包含染色质可及性、CpG甲基化和染色质构象信息的长片段进行测序,在单分子分辨率上将染色质可及性和CpG甲基化映射到基因组空间结构,同时解析基因组构象。
基因组在细胞内具有三维结构,这使得基因组区域的可及性具有类似的空间复杂性,其表观基因组信息也具有复杂的空间排列。因此,有必要使用精密的工具来深入研究染色质可及性,从而全面理解染色质活化和基因的相互作用。
值得注意的是,仅依靠解析染色质的可及性并不能完全捕捉其空间排列的复杂性。染色体构象捕获技术,例如Hi-C、SPRITE、CHIAPET和pore-C,已被广泛用于绘制全基因组染色质结构,但是这些技术大多无法捕获表观基因组信息,而染色质区域和甲基化标记信息的缺失限制了利用多组学方法研究全基因组结构的可能性。因此,利用多组学信息重建基因组组织结构有助于进一步理解转录的相互调控作用,并促进DNA相互作用的研究。
近日,华大基因研究团队在预印本bioRxiv发表了题为“Spatial chromatin accessibility sequencing resolves high-order spatial interactions of epigenomic markers”的文章,报道了基于甲基化标记和邻近连接的空间染色质可及性测序(Spatial chromatin accessibility sequencing, SCA-seq)。SCA-seq利用纳米孔技术对包含染色质可及性、CpG甲基化和染色质构象信息的长片段进行测序,在单分子分辨率上将染色质可及性和CpG甲基化映射到基因组空间结构,同时解析基因组构象。基于该技术,研究团队揭示了在空间相互作用中存在异质染色质可及性,表明了复杂的基因组调控。SCA-seq有助于促进基因组空间组织的多组学研究。

主要研究内容
SCA-seq技术原理
研究团队旨在使用SCA-seq研究染色质空间密度,同时解决基因组组织和染色质可及性(图1.1)。细胞固定后,研究团队使用甲基转移酶(EcoGII或M.CviPI)人工标记染色质可及性区域(图1.2-3)。在染色质可及性信息被保存为m6A或GpC甲基化标记后,使用染色质构象捕获方案进行消化和连接步骤,利用邻近连接将多个在三维空间中线性距离相互接近的DNA片段连接在一起(图1.4),并通过优化DNA提取方法获得更纯的大片段DNA(图1.5)。随后,利用纳米孔方法对携带染色质可及性信息、甲基化标记和三维构象信息的DNA片段进行测序及分析。(图1)与其它技术相比,SCA-seq保存了更多的多组学信息,例如CpG甲基化表观遗传标记、染色质可及性和高阶染色质构象数据(图2)。
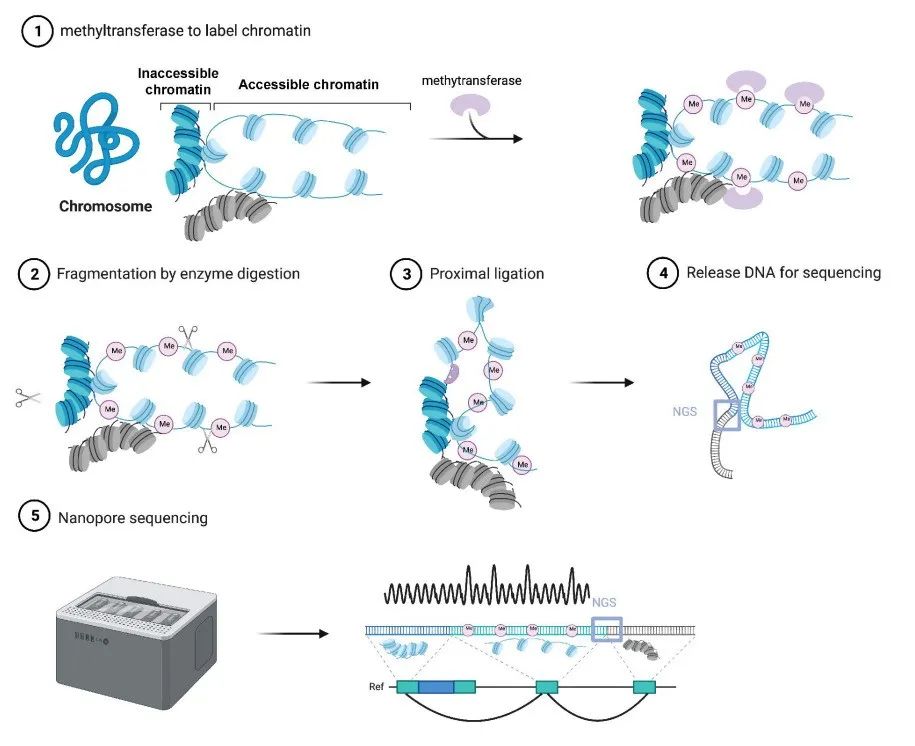
图1. SCA-seq技术原理及测序步骤。
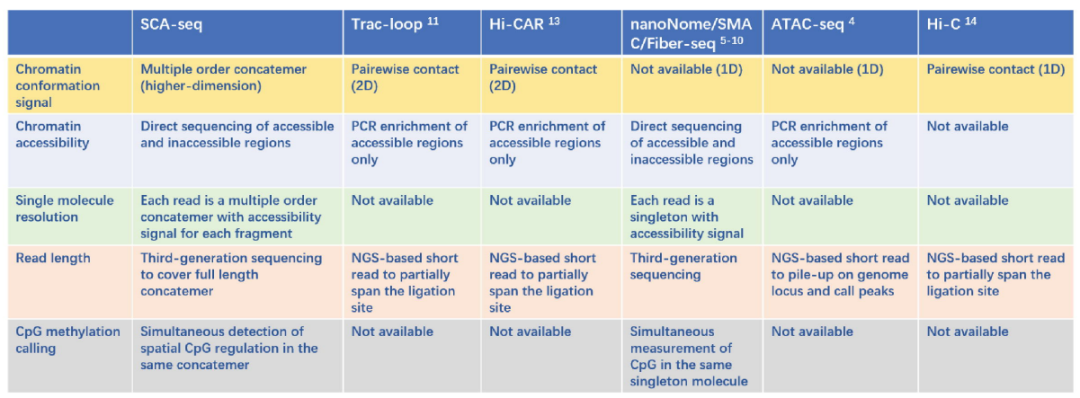
图2. SCA-seq与其它技术比较。
识别染色质可及性和天然甲基化标记
接下来,研究团队评估了SCA-seq同时揭示天然甲基化和染色质可及性的潜力。
首先验证了SCA-seq中甲基化检测和甲基转移酶标记的准确性,通过对HEK293细胞的测序数据进行初始质量控制,最终生成了129.94 Gb(36.9×)的测序数据,N50长度为4446bp。为了从纳米孔数据中获取甲基化信息,研究团队采用Nanopolish和cpggpc检测模块,取得了相当大的成功(AUC CpG = 0.908, GpC = 0.984)。利用金标准全基因组亚硫酸酯测序的进一步分析结果与Nanopolish的结果高度相关(图3),支持SCA-seq甲基化检测的准确性。此外,经验证,SCA-seq使用的生物信息学流程能够检测天然CpG甲基化和人工标记的GpC甲基化。
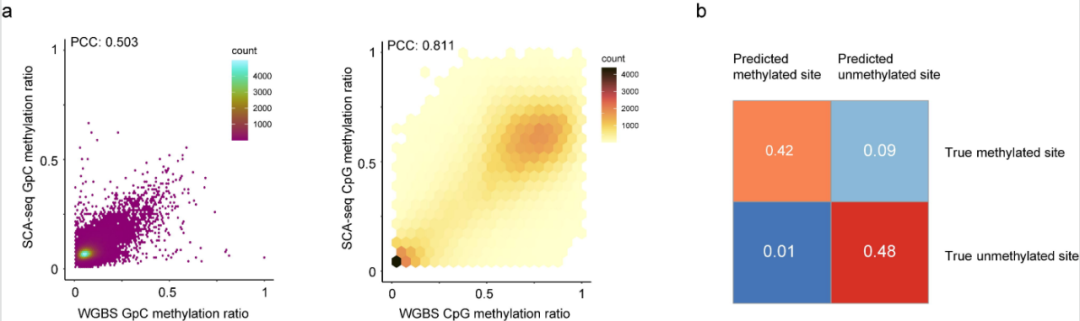
图3.SCA-seq甲基化检测的可行性。
由于ATAC-seq和DNase-seq是检测染色质可及性的金标准,因此,研究人员将SCA-seq与ATAC seq/DNase-seq进行了全局和局部比较。在SCA-seq的全基因组数据中分析得到87991个可及性峰,其中80%与ATAC-seq或DNase-seq中的峰重叠(图4)。在局部比较中,SCA-seq也显示与ATAC-seq识别的峰相似的峰型。
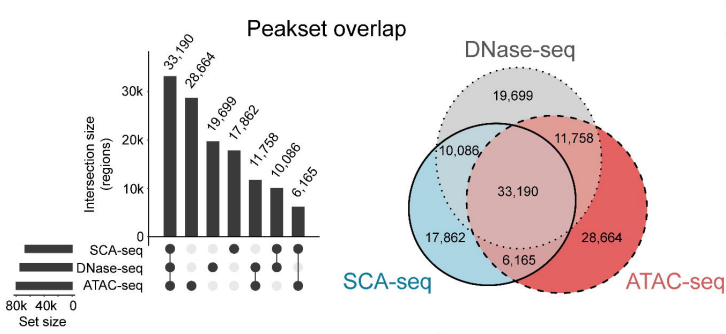
图4. 检测染色质可及性,SCA-seq、ATAC-seq和DNA-seq之间峰重叠的维恩图。
此外,研究团队还计算预测了CCCTC结合因子(CTCF)的结合位点,发现局部CpG甲基化水平下降,CTCF结合位点周围的GpC可及性增加(图5)。SCA-seq在基因组水平上可靠地检测到了染色质可及性。
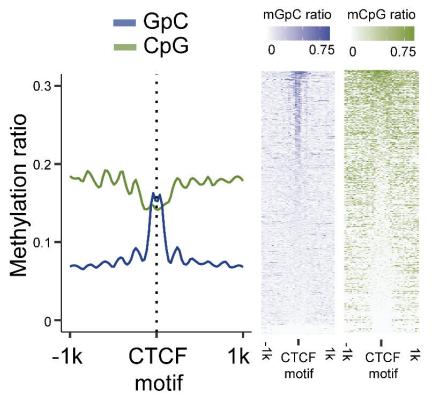
图5.检测染色质可及性,CTCF基序位点图。
捕获高阶染色质结构和CTCF足迹
除了甲基化信息外,SCA-seq还保存了基因组的空间结构。研究团队比较了SCA-seq与金标准Hi-C在解析基因组空间组织能力方面的差异。结果发现,SCA-seq显示的基因组组织类似于原位Hi-C检测到的。通过SCA-seq和Hi-C获得的交互热图、环路、TAD边界和A/B区并行可视化显示了等效的基因组组织。以上结果表明,SCA-seq成功地解析了染色质相互作用的多重性质。
利用SCA-seq观察到染色质构象具有特定的结合模式,例如CTCF结合模式。研究发现,与非CTCF结合相比,CTCF结合导致了更高阶和更多的远端相互作用,这表明CTCF结合促进了更复杂结构的形成。
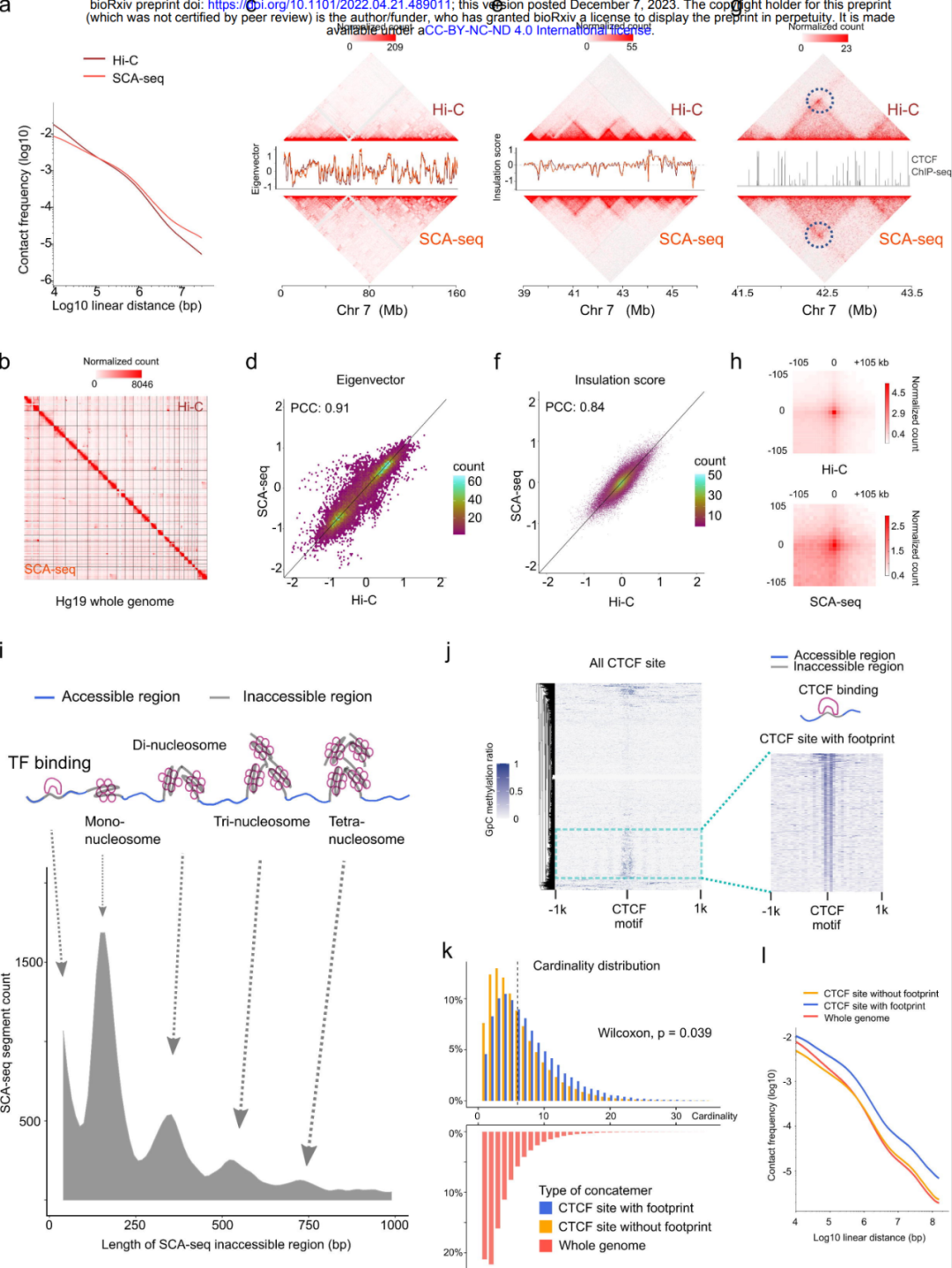
图6. SCA-seq能够捕获高阶染色质结构和CTCF足迹。
揭示可及和不可及染色质区域的空间相互作用
由于不同细胞的基因组组织具有高度异质性,因此,染色质相互作用状态分析主要依赖于单分子模式,这需要很高的敏感性和特异性。分析发现,可及和不可及的DNA在SCA-seq数据中连接在一起,表明了空间相邻DNA区域可及性的异质性。此外,62.2%的基因组串联中既有可及片段,也有不可及片段。
研究发现,与不可及的串联子相比,杂合的串联子包含更多的增强子和启动子元件,提示了串联子类型与转录调控程度之间的关系。SCA-seq可及性信号提示空间相互作用和染色质可及性可能协同调控基因表达水平。
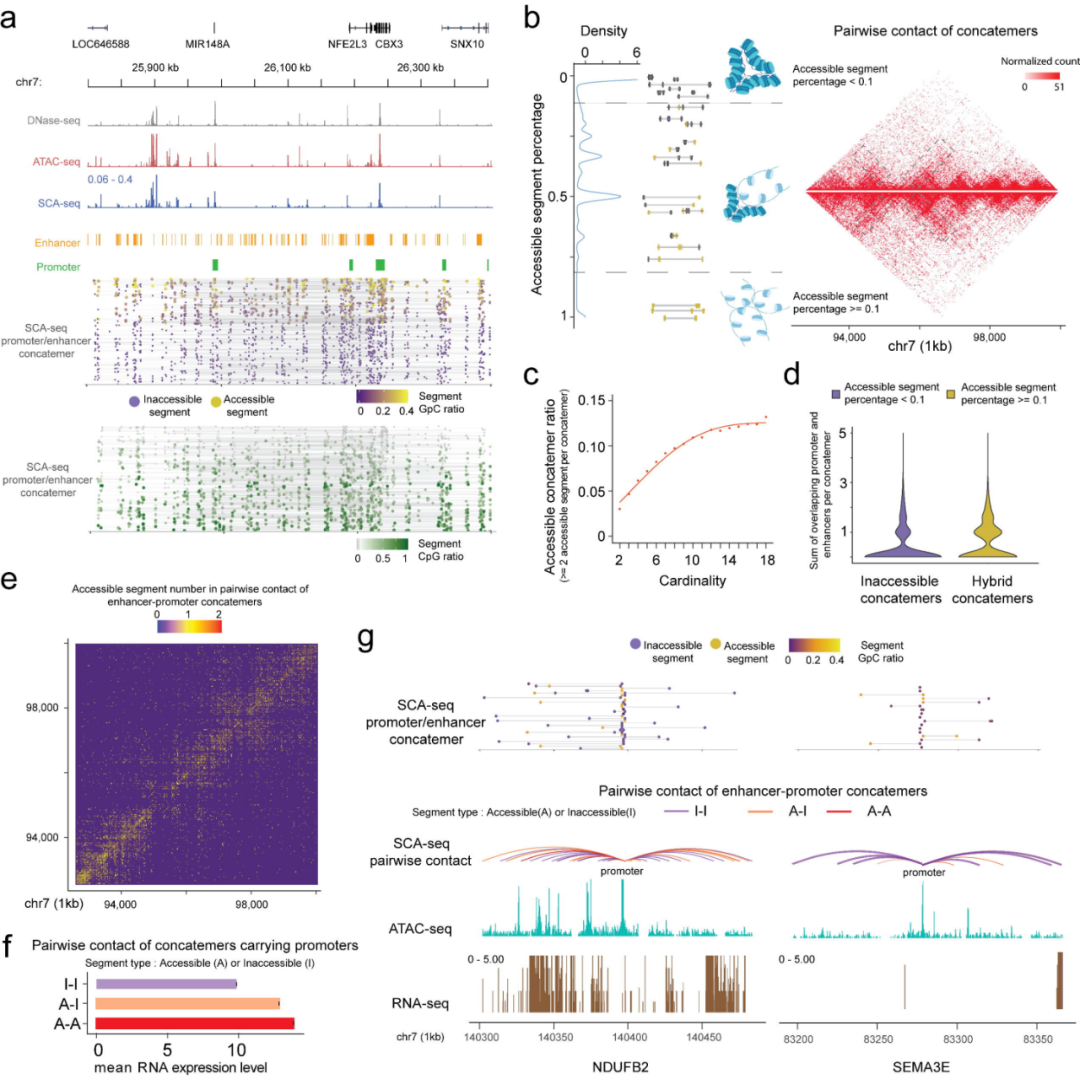
图7. SCA-seq揭示空间染色质可及性。
揭示与“孤儿”CpG岛空间接触的CpG甲基化
CpG岛(CGI)是脊椎动物基因组的普遍特征,与约50%的基因启动子相关(pCGI)。另有成千上万的“孤儿”CGI (oCGIs),它们与启动子和增强子之间的距离较远,几乎不为人所知。在显示高阶相互作用和CpG甲基化的SCA-seq数据中,研究发现7号染色体上的oCGIs有76418个reads重叠,并且大多数oCGIs通常在空间上靠近CTCF结合基序和活性组蛋白标记物。分析显示,oCGIs能够将增强子和CTCF基序连接起来,并可与启动子进行作用,拓展了人们对oCGIs调控功能的理解。
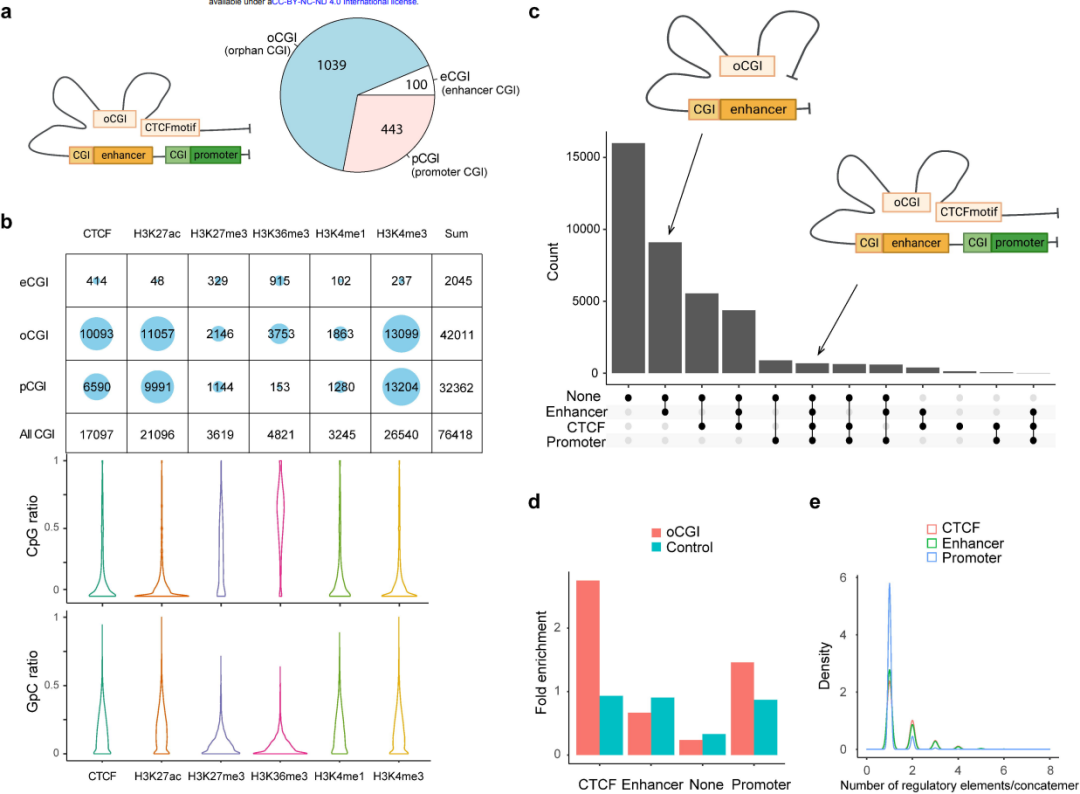
图8. CpG甲基化与CpG岛不同类型的空间联系。
结 语
目前大多数表观遗传学研究仅在水平维度描述了甲基化标记、染色质可及性区域和组蛋白修饰,相关空间表观基因组信息较少。该研究团队报道了新开发的SCA-seq,将传统染色质可及性的概念扩展到高维空间,以单分子分辨率同时分析DNA甲基化、染色质可及性、转录因子结合和染色质构象。通过SCA-seq,研究发现基因组空间结构保持了染色质的非均匀可及性,表明基因组在3D空间中具有复杂的调控。
同时,SCA-seq还可以作为一种多组学工具,在三维空间中解析基因组的可及性位置,促进染色质构象区域亚群的发现。因此,SCA-seq可能为更详细地探索动态基因组结构铺平了道路。
论文原文:
Yeming Xie, Fengying Ruan, Yaning Li, et al., Spatial chromatin accessibility sequencing resolves high-order spatial interactions of epigenomic markers. bioRxiv (2023).
https://www.biorxiv.org/content/10.1101/2022.04.21.489011v4.full.pdf
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#测序# #空间染色质# #SCA-seq#
35