NAR:北京大学汤富酬团队利用单精子长读段基因组测序进行高精度单倍型分型
2023-07-18 iNature iNature 发表于上海

该研究开发了一种基于长读长的单精子基因组测序方法和相应的数据分析管道,可以准确地识别单个精子中的交叉事件和染色体水平的非整倍体,并有效地检测单个精子细胞中的SVs。
尽管在没有亲本数据的情况下,利用长读长的基因组测序可以实现单倍体的定位,但是可靠的染色体尺度的分期仍然是一个巨大的挑战。鉴于精子是天然的单倍体细胞,单精子基因组测序可以提供全染色体的相位信号。由于读长的限制,目前基于短读长的单精子基因组测序方法只能实现SNP单倍型分型,难以检测和分型复杂基因组区域的结构变异( Structural Variations,SVs )。
2023年6月23日,北京大学汤富酬团队在Nucleic Acids Research 上在线发表题为“Long-read-based single sperm genome sequencing for chromosome-wide haplotype phasing of both SNPs and SVs”的研究论文,该研究开发了一种基于长读长的单精子基因组测序方法和相应的数据分析管道,可以准确地识别单个精子中的交叉事件和染色体水平的非整倍体,并有效地检测单个精子细胞中的SVs。
重要的是,在没有亲本基因组信息的情况下,该方法可以在整个染色体尺度上准确地对来自雄性个体的杂合SVs和SNPs进行从头阶段划分。利用100个单个精子细胞对SVs进行分期的准确率高达98.59 %,对SNPs进行分期的准确率高达99.95 %。此外,该方法能够可靠地推断出单倍型分辨的STRs / VNTRs在单个精子细胞中的重复扩展,为研究哺乳动物单倍型相关的遗传学提供了新的机会。

配子是天然的单倍体细胞,其携带的遗传信息可被后代遗传,使配子成为遗传学研究的理想对象。第一个单精子基因组测序技术是在十年前发展起来的,此后测序方法的不断改进为单配子研究提供了新的见解。不同的单精子测序方法具有独特的特点,如在短读长测序平台上采用微滴或组合索引( SCI )策略的高通量单精子基因组测序方法或通过对单个精子进行高深度测序实现的高基因组覆盖率方法。尽管这些方法各不相同,但共同的生物学问题都是用单个精子细胞来解决的,并且集中在三个领域:减数分裂和交叉的研究、精子基因组不稳定性和单倍型的阶段化。
利用短读技术对单个配子进行测序,能够使研究减数分裂表型变得简单和准确,而不需要在亲属间共享信息的基因组片段,也不需要分析群体中的连锁不平衡模式。此外,牛津纳米孔技术( Oxford Nanopore Technologies,ONT )、太平洋生物科学( Pacific Biosciences,PacBio )或Linked - Read测序等长读长测序技术也被开发用于大批量精子基因组测序,主要用于提高交叉点的检测分辨率。这些现有的测序方法为减数分裂研究提供了极大的技术支持。
目前,已经有许多研究利用短读技术研究精子细胞中单核苷酸变异( SNVs )和CNVs之间的关联及其与疾病的潜在关系。然而,SVs引起的个体间基因组差异估计比SNVs引起的个体间基因组差异高3 ~ 10倍。在减数分裂中,同源染色体对通过同源重组和交叉交换进行遗传物质的物理交换,这需要DNA双链断裂和修复过程。这些修复事件可以导致基因组的变化,从SNV到结构和拷贝数的变化,其中一些可能会对后代产生重大的影响。然而,由于短读技术难以检测复杂基因组区域的遗传变化,且检测SVs的假阳性率较高,因此对精子中SVs的研究受到了限制。
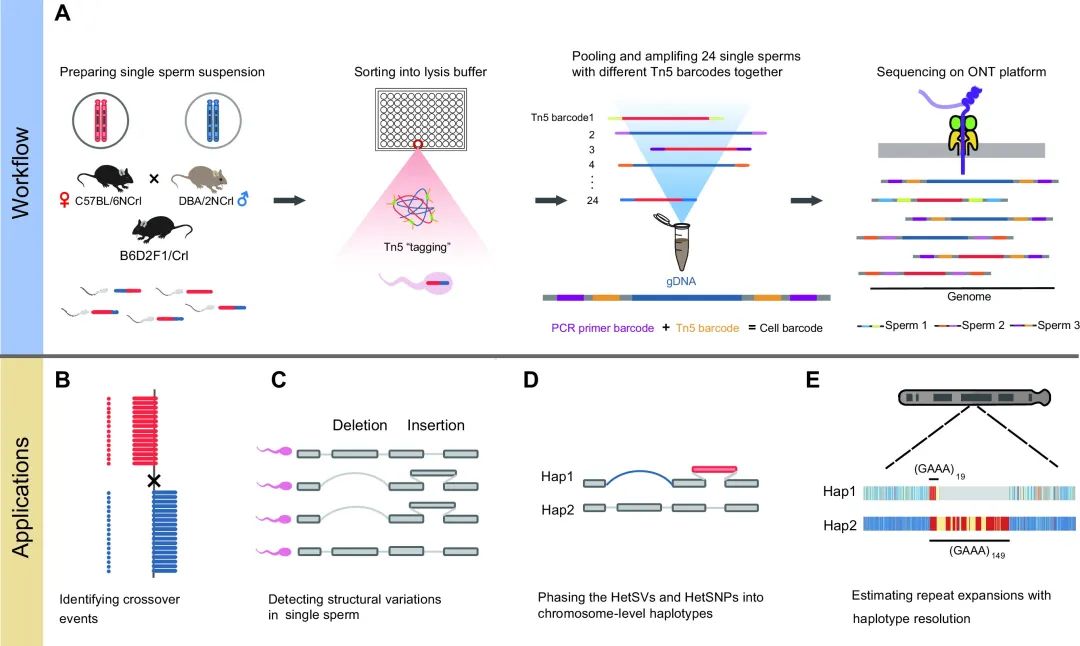
基于长读的单精子基因组测序工作流程(图源自Nucleic Acids Research )
事实上,SVs的研究是有价值的,尤其是对于重复型SVs ( SVs的序列中含有重复元件)。重复元件( Repetitive element,RE )广泛存在于基因组中,通常可分为串联重复序列和转座元件( Transposable element)两种类型,两者对基因组稳定性具有重要影响,并已被证明在调控基因表达、RNA剪接和DNA甲基化等方面发挥重要作用。重要的是,超过30种孟德尔疾病是由串联重复序列通过一系列分子机制引起的。由于缺乏对配子基因组中SVs的可靠研究,可能会错失研究不育或遗传疾病的机会。
为了应对这些挑战,该研究开发了一种基于长读长的单精子基因组测序方法和相应的数据分析流程。研究人员对F1代杂交小鼠B6D2F1 / Crl [ BDF1 ] (雌性小鼠C57BL/6NCrl小鼠[ B6 ] ×雄性小鼠DBA / 2NCrl [ DBA ])的精子细胞进行测序,设计了24种不同条形码的Tn5酶。通过组合后续扩增引物的不同条形码进行单精子基因组测序,可以实现较高的扩增效率。通过长读长测序,研究人员能够准确地检测单个精子细胞中的结构变异( SVs )。更重要的是,基于长读长的单精子基因组测序实现了全染色体分期和单体型特异性性质DNA特征的测量。长读长也使识别单倍型分辨的串联重复序列( STRs / VNTRs )在单个精子细胞中的扩增成为可能。
北京大学生物医学前沿创新中心、北京大学生命科学学院博士谢昊伶、北京大学前沿交叉学科研究院博士生李文以及北京大学生命科学学院博士生郭雨晴为该论文的并列第一作者。北京大学生物医学前沿创新中心汤富酬教授为该论文的通讯作者。该研究项目得到了北京大学生物医学前沿创新中心、北京未来基因诊断高精尖创新中心、北大-清华生命科学联合中心、北京昌平实验室、北京市科技委的支持。
参考信息:
https://doi.org/10.1093/nar/gkad532
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言