韩国糖原贮积病Ⅵ型患者的临床和遗传图谱
2023-08-26 田医生 MedSci原创 发表于上海

GSD基因Panel检测是确诊GSD-Ⅵ的有效诊断工具,所有GSD-Ⅵ患者均存在遗传异质性,经过长期随访,治疗后患者的肝酶升高、高甘油三酯血症和身高Z评分可得到改善。
糖原累积病(glycogen storage disease,GSD)是糖原代谢过程中各种酶缺陷导致的一组先天性遗传代谢性疾病,以糖原在全身各器官组织中过度沉积为特征,主要累及肝脏或(和)肌肉,是一类较常见的代谢性疾病,发病率1:20000~43000。根据受累器官,又被分为肝糖原累积病和肌糖原累积病。肝糖原累积病包括Ⅰ、Ⅲ、Ⅳ、Ⅵ、Ⅸ、Ⅺ及O型等,约占GSD总体发病率的80%。
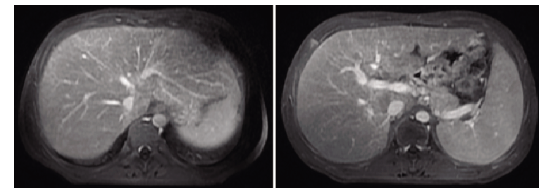
其中糖原贮积病Ⅵ型(glycogen storage disease type Ⅵ,GSD-Ⅵ)呈常染色体隐性遗传,是由于肝糖原磷酸化酶缺乏而导致糖原异常蓄积的代谢障碍性疾病,活产儿发病率为1:60000~85000,大部分患者于婴幼儿时期因肝大和生长迟滞就诊,主要临床表现有:肝大、腹部膨隆、生长迟滞、空腹低血糖、肝转氨酶升高等,但智力一般正常,预后相对良好。
GSD-Ⅵ是由位于染色体14q21-q22上的PYGL基因突变引起的先天性糖原代谢错误,PYGL基因是唯一致病基因,包含20个外显子,编码糖原磷酸化酶L蛋白,该蛋白为同源二聚体蛋白,通过催化α-1,4-葡萄糖苷键断裂使肝糖原释放葡萄糖-1-磷酸参与糖原分解。
GSD-Ⅵ诊断的金标准是肝活检酶检测,但它具有侵入性,因此最新指南建议首先进行分子诊断。此外,其他类型的GSD也存在肝肿大、低血糖等临床表现。特别是GSD-Ⅵ的临床表型与GSD-Ⅸ的临床表型重叠,只能通过分子诊断来区分。
在韩国,尚没有研究报告GSD-Ⅵ患者的临床特征和PYGL基因变异。Jong Woo Hahn等人的团队研究了韩国GSD-Ⅵ患者的临床特征、肝脏组织学、PYGL变异和长期预后。
团队回顾性分析了从2002年1月至2022年11月首尔国立大学医院使用基因Panel检测诊断为GSD-Ⅵ的患者。
共有5例患者纳入研究。发病年龄18~30个月(中位年龄21个月),当前年龄3.7~17岁(中位年龄11岁)。所有患者均表现为肝肿大、转氨酶活性升高和高甘油三酯血症。高胆固醇血症和空腹低血糖分别发生在60%和40%的患者中。共鉴定出10个PYGL变异,其中6个为新变异:5个错义突变和1个移码突变。所有患者都接受了高蛋白饮食治疗,其中四名患者也接受了玉米淀粉治疗。
团队提出GSD基因Panel检测是确诊GSD-Ⅵ的有效诊断工具,所有GSD-Ⅵ患者均存在遗传异质性,经过长期随访,治疗后患者的肝酶升高、高甘油三酯血症和身高Z评分可得到改善。
参考文献:
Hahn Jong Woo,Lee Heerah,Seong Moon Woo et al. Clinical and genetic spectrum of GSD type 6 in Korea.[J] .Orphanet J Rare Dis, 2023, 18: 132.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言