
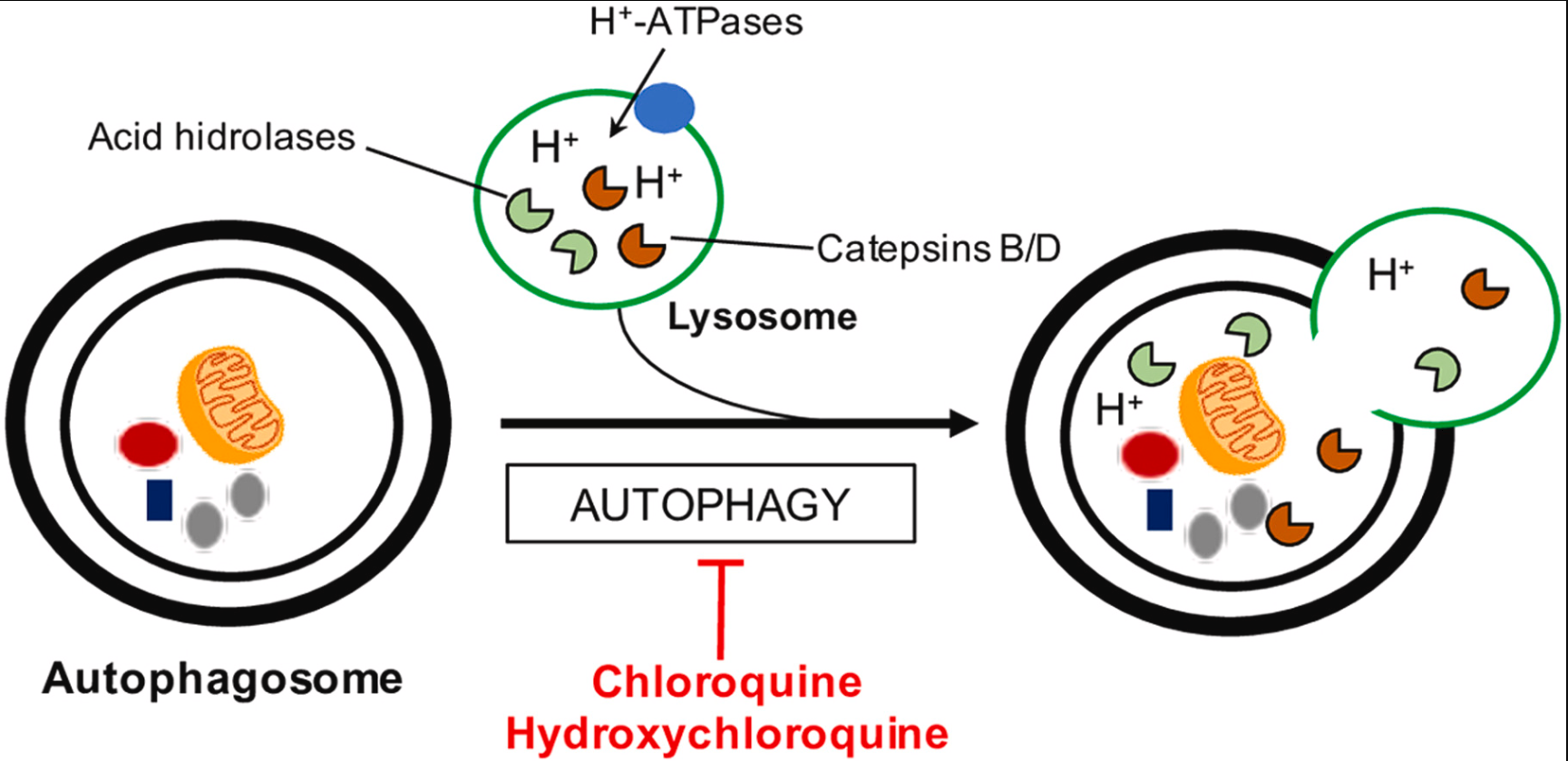
细胞死亡在生物体的发育和稳态维持中起着关键作用。不同形式的细胞死亡,包括凋亡(Apoptosis)、坏死性凋亡(Necroptosis)、焦亡(Pyroptosis)和铁死亡(Ferroptosis),不仅在清除受损或过时的细胞中发挥作用,也在抑制病原体扩散中扮演重要角色。24年1月,Cell杂志刊文综述了各种细胞死亡路径的信号机制,探讨这些细胞死亡过程的异常激活或抑制如何导致疾病,并讨论了针对细胞死亡失衡状态的现有和潜在治疗方法。

细胞死亡可以是非溶解性的,且在免疫上相对沉默(凋亡),或者是溶解性的且促炎症的(坏死)。在多细胞生物中,基因程序化的细胞死亡被认为是维持稳态的重要支柱,但即使是单细胞生物也可以通过细胞死亡来抵御病原体或为适应营养匮乏而限制群落大小。非溶解性的细胞死亡程序最初是基于其形态学特征来定义的,这些特征包括细胞质空泡化、核浓缩和细胞膜起泡。其背后的分子机制在三个不同领域的多年研究之后才逐渐清晰。
首先是认识到在遗传上易于操作的秀丽隐杆线虫(Caenorhabditis elegans)中某些细胞可重复地经历凋亡。随后的遗传筛选识别出了细胞死亡的介导者(egl-1、ced-3和ced-4)和细胞死亡的抑制因子(ced-9),尽管当时它们还不能被赋予可识别的生化活性。关于ced-3功能的认识来自于对哺乳动物促炎症白细胞介素-1β(IL-1β)成熟的蛋白酶的鉴定。CED-3与这种现在被称为半胱天冬酶-1(caspase-1)的IL-1β转化酶之间的同源性促使人们认识到CED-3 是一种蛋白酶,并且秀丽隐杆线虫中的程序化细胞死亡是由蛋白水解驱动的。
ced-9基因在哺乳动物中有一个功能对应物,即B细胞淋巴瘤基因2(BCL-2),这是一种在人类滤泡性淋巴瘤中通过染色体易位激活的原癌基因。与许多其他原癌基因不同,BCL-2并不诱导增殖,而是抑制细胞死亡,并且能够在线虫中功能性地替代ced-9。在这些关键发现之后,进一步鉴定出了额外的哺乳动物半胱天冬酶(Caspase,半胱氨酸依赖的天冬氨酸定向蛋白酶)。此外,BCL-2被发现是控制Caspase激活的一个更大家族的成员,这个家族被称为内源性凋亡途径的裁决者。在外源性凋亡途径的形式中,出现了更多的复杂性,以及病原体对死亡途径成分的征用。对受感染细胞的研究显示,某些形式的坏死(即细胞破裂并释放促炎症内容物)也是基因程序化的。这些细胞死亡途径被称为焦亡(pyoptosis)和坏死性凋亡(necroptosis)。拥有多种死亡方式是对抗寻求保持其复制生态位的病原体的有效寄宿策略。
在成年人中估计每天有惊人的10^11个细胞经历程序化细胞死亡,这相当于我们一年内整个体重的细胞。通过细胞增殖,身体维持现状,同时排除老化、功能失调、受感染或突变的细胞。因此,扰乱细胞增殖与细胞死亡之间的稳态平衡将导致机体功能失调。过少的细胞死亡会导致过度增殖疾病,如癌症,而过多的细胞死亡则导致退行性疾病,如神经退行性疾病。
在这里,本文主要基于当前对各种细胞死亡途径的主流观点,由于研究众多,有的令人困惑,甚至矛盾。为了避免遗漏某些进展,我们主要选择那些通过生物化学和遗传学相结合的方法经过考验的观点和证据,相对更能经受住时间的考验。
内源性凋亡——从线虫到哺乳动物
在秀丽隐杆线虫(C. elegans)中,许多内源性凋亡信号传导在哺乳动物中也有保留,但哺乳动物的细胞死亡途径更为复杂(见图1)。相似之处包括使用支架蛋白来协调半胱天冬酶(caspase)的激活。在C. elegans中,CED-4担当此功能,用于激活半胱天冬酶CED-3,而在哺乳动物中,凋亡肽酶激活因子1(Apoptotic peptidase activating factor 1,APAF-1)介导caspase-9的聚合。这一过程在BCL-2同源区3(BH3)-仅蛋白(BH3-only proteins)(如C. elegans中的EGL-1;哺乳动物中的细胞凋亡相关BCL-2拮抗剂(BAD),BH3相互作用域死亡拮抗剂(BID),BCL-2相互作用杀手(BIK),BCL-2相互作用的细胞死亡介质(BIM),BCL-2修饰因子(BMF),harakiri(HRK),NOXA(也称为12-肉豆蔻酸-13-醋酸诱导蛋白1)、以及p53上调的凋亡调节剂(PUMA))与BCL-2蛋白家族的促存活成员(C. elegans中的CED-9;哺乳动物中的BCL-2、BCL-XL、髓细胞白血病基因1(MCL-1)、BCL-W以及胎儿肝中表达的BCL-2相关基因(BFL-1;在小鼠中称为A1))结合,从而抑制它们。这些BH3-仅蛋白(BH3-only proteins)的表达在发育信号或应激刺激(例如,营养剥夺、DNA损伤和内质网(ER)应激)的作用下增加,通过多种转录和翻译后机制实现。
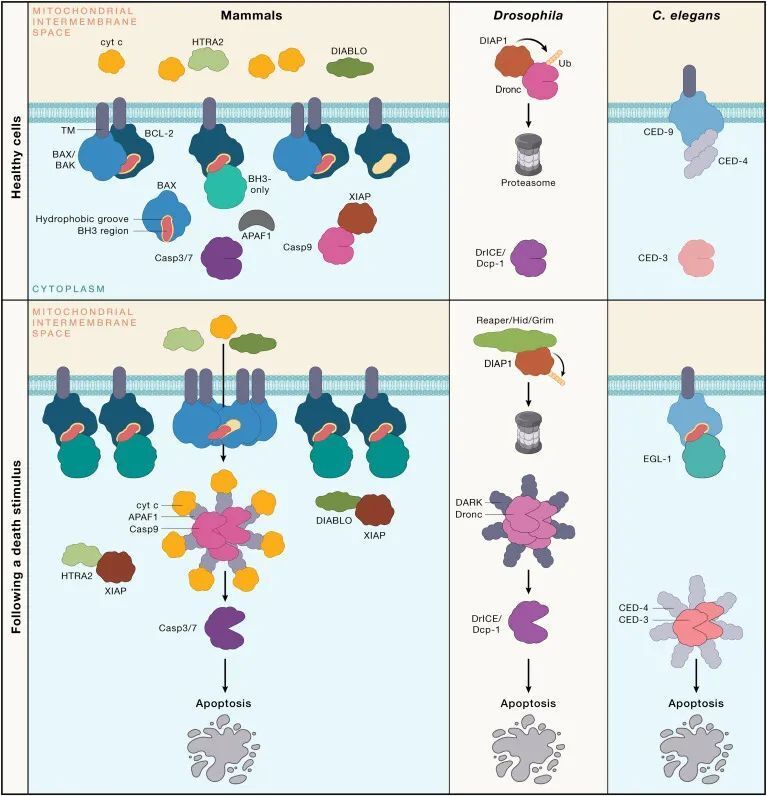
图1 - 哺乳动物内源性凋亡信号传导途径与秀丽隐杆线虫和果蝇的凋亡信号传导对比(Credit: Cell)
在C. elegans中,CED-9通过阻止CED-4激活caspase CED-3来维持细胞存活。相比之下,哺乳动物中促存活的BCL-2家族蛋白通过限制它们的促凋亡成员,即BCL-2相关X蛋白(BAX)和BCL-2拮抗剂/杀手1(BAK),来防止caspase的激活。未受抑制的BAX和/或BAK聚合,并导致线粒体外膜渗透(Mitochondrial outer membrane permeabilization,MOMP),从而释放促进凋亡的线粒体因子进入细胞质。线粒体释放的细胞色素c与adaptor APAF-1相互作用,促进凋亡体复合体的组装,有助于起始caspase,即caspase-9的二聚化和激活。线粒体释放的DIABLO(也称为线粒体衍生的第二凋亡激活因子[SMAC])或HtrA丝氨酸蛋白酶2(HTRA2)阻止X连锁的凋亡蛋白抑制剂(XIAP)对caspase的抑制。虽然C. elegans没有类似BAK或BAX的蛋白,但其他线虫却有。因此,C. elegans可能是进化中的一个特例,因为它通过与CED-4的结合,以更直接的方式抑制caspase的激活,从而放弃了对凋亡的线粒体调控。或者,被BH3-only protein EGL-1结合的CED-9可能像BAX或BAK一样发挥功能。有证据表明CED-9与线粒体外膜相关,但EGL-1的亚细胞定位不太清楚。
哺乳动物的caspase-9通过蛋白水解激活所谓的执行者caspase(caspase-3、-7和可能的-6),导致数百种细胞蛋白的切割。这些切割事件中的一些有助于拆解细胞,而其他则激活促进吞噬凋亡细胞及其膜包裹的碎片(称为凋亡体,apoptotic bodies)的过程。前者的一个例子是caspase-3或-7对caspase激活的脱氧核糖核酸酶抑制剂(ICAD,inhibitor of caspase-activated deoxyribonuclease)的切割,这释放了caspase激活的脱氧核糖核酸酶(CAD,caspase-activated deoxyribonuclease)以介导染色体DNA的核间切割。后者的一个例子是对XK相关蛋白8(XKR8,XK related 8)的蛋白水解激活,导致磷脂酰丝氨酸在细胞表面显示为“吃我”的信号,以吸引吞噬细胞。
吞噬作用对凋亡的非炎症性质很重要。如果凋亡细胞未被“吞噬”,它们最终可能发展出膜损伤。细胞内损伤相关分子模式(DAMPs, damage-associated molecular patterns)的释放向邻近细胞发出“警报”,触发促炎症反应。DAMPs包括DNA、ATP和蛋白质,如高迁移率族蛋白盒1(HMGB1, high mobility group box 1)和IL-1α。调节凋亡细胞的吞噬作用的过程在进化过程中得到了保留,凸显了它们对正常发育和成年组织稳态的重要性。
果蝇(Drosophila melanogaster)是另一种用于研究程序性细胞死亡(programmed cell death)的模式生物。在果蝇中,Debcl、Buffy和Sayonara属于BCL-2蛋白家族(BCL-2 protein family),但它们在程序性细胞死亡中的作用比其哺乳动物和秀丽隐杆线虫(C. elegans)的对应物有限。果蝇的半胱天冬酶(caspases)主要受到细胞凋亡抑制蛋白(IAPs, inhibitor of apoptosis proteins)的控制(见图1)。果蝇中程序性细胞死亡的触发是由IAP抑制剂Reaper、Hid和Grim的诱导表达引起的。哺乳动物的IAPs在抑制细胞死亡中也扮演重要角色,但XIAP是唯一直接针对caspase的哺乳动物IAP,它抑制caspase-3、-7和-9。
BCL-2蛋白家族
BCL-2家族的成员可以分为三个亚组:促凋亡的BH3-only protein、促生存蛋白和促凋亡效应蛋白。哺乳动物的促生存蛋白具有四个BH区域(BH regions)、一个C末端跨膜域(TM domain, transmembrane domain)和一个疏水表面凹槽,用于介导与BCL-2家族两个促凋亡亚组的BH3域的相互作用。效应蛋白BAX、BAK和与BCL-2相关的卵巢杀手蛋白(BCL-2-related ovarian killer,BOK)的结构与促生存蛋白非常相似。它们也具有四个BH区域(由一个保守序列基序定义的BH4域)、一个TM域和一个BH3结合表面凹槽。相比之下,许多BH3-only protein在未与促生存家族成员结合时是未结构化的(unstructured)。
促凋亡效应蛋白BAX、BAK和BOK
大多数健康细胞都表达可检测量的BAX和BAK,尽管有些细胞表达其中之一的量要多得多。缺乏BAX的小鼠表现出雄性不育和轻微的脾肿大,而缺乏BAK的小鼠基本正常。然而,BAX和BAK的联合缺失通常产生严重的颅面异常,这在出生时是致命的。这种表型在缺乏BOK的小鼠中会加剧。与BAX和BAK不同的是,BOK似乎不受促生存BCL-2蛋白的抑制。BOK的丰度和活性反而受到位于内质网(ER)的泛素连接酶gp78的抑制,gp78将BOK定位为蛋白酶体降解。来自Bax−/− Bak−/−或Bax−/− Bak−/− Bok−/−小鼠的细胞对所有测试的内源性凋亡刺激都极度抵抗,这表明BAX和BAK在凋亡中具有很大的重叠功能,作为必需的凋亡效应蛋白,而BOK的作用是辅助的。尽管它们具有广泛的功能重叠,BAX和BAK之间还是有显著差异。尽管两者都与线粒体外膜上的促生存BCL-2蛋白相互作用,但健康细胞中的大多数BAX是细胞质的,其跨膜结构域嵌入其疏水凹槽内。BAX激活时跨膜域是如何被置换的目前还不太清楚。BAX和BAK的另一个区别是,BCL-2被认为主要抑制BAX,而MCL-1抑制BAK,但BCL-XL可以有效地抑制这两种蛋白。有趣的是,电压依赖性阴离子通道2(VDAC2, voltage-dependent anion channel 2)允许BAX定位到外线粒体膜上杀死细胞,但相反,它可以抑制BAK的激活。尽管经过多年的深入研究,BAX或BAK形成的介导线粒体外膜通透性改变(MOMP)的孔的结构仍然是个谜。
BH3-only proteins
BIM、PUMA和被蛋白水解裂解的BID(称为截短的BID或tBID)与所有促生存BCL-2蛋白高亲和力结合,因此是强有力的凋亡启动因子。其他的BH3-only protein则以更选择性的方式结合。NOXA仅与MCL-1和A1/BFL-1结合,而BAD、BIK和HRK主要与BCL-XL结合,程度较小的与BCL-2和BCL-W结合。因此,这些BH3-only protein往往是较弱的杀手,因为它们只抑制保护细胞的部分促生存BCL-2蛋白。特定的BH3-only protein,尤其是BIM、PUMA和tBID,也可能通过与BAX和BAK直接相互作用来触发凋亡。然而,对缺乏所有BH3-only蛋白的细胞系的分析表明,这些相互作用对启动凋亡并非必需。当保护细胞的促生存BCL-2蛋白通过遗传手段移除或用小分子BH3模拟物抑制时,细胞仍然经历BAX或BAK介导的凋亡。这些发现总体上与一种模型一致,即BAX和BAK需要被促生存BCL-2蛋白抑制以维持细胞活性。当促生存BCL-2蛋白被BH3-only protein或蛋白酶体降解(下文描述)中和时,凋亡就会启动。
启动凋亡的BH3-only protein取决于应激刺激和细胞类型。BIM在免疫耐受中起主要作用,通过清除自反应性B细胞和T细胞。它还通过移除免疫反应后不再需要的B细胞和T细胞,帮助免疫细胞的稳态。BIM和PUMA促进由生长因子剥夺、钙通量失调、糖皮质激素或内质网(ER)应激引发的凋亡。转录因子和肿瘤抑制因子p53(在小鼠中也称为转化相关蛋白53 [TRP53] (transformation related protein 53),在人类中称为肿瘤蛋白p53 [TP53] (tumor protein p53))通过诱导Puma和Noxa的表达促进凋亡。BID通过caspase-8在外源性凋亡途径(下文描述)或通过细胞毒性T细胞或自然杀伤细胞释放的颗粒酶B (granzyme B)裂解而激活,后者通过穿孔素孔进入细胞。针对基因定位的小鼠的分析表明,BMF、BAD、BIK和HRK在启动凋亡中的作用不太突出。
Pro-survival BCL-2 proteins
所有促生存BCL-2蛋白(pro-survival BCL-2 proteins)都定位于线粒体外膜的外侧,但BCL-2也存在于内质网和核膜上。BCL-XL的相当部分是细胞质的。BCL-2、BCL-XL和BCL-W相对稳定,需要数小时才能转换,而MCL-1和A1/BFL-1是不稳定的蛋白,它们的半衰期以分钟而非小时计算,因为它们被泛素化并定向蛋白酶体降解。因此,当RNA或蛋白质合成停止时,MCL-1和A1/BFL-1的水平会急剧下降,依赖它们的细胞将死亡。许多病毒也产生促生存BCL-2同源物或结构类似物,强调内源性凋亡在消除病毒感染细胞中的重要作用。
在小鼠中的遗传研究确定了不同促生存BCL-2蛋白的关键功能。A1的丧失只引起选择性造血细胞亚群的轻微缺陷。缺乏BCL-W的小鼠也基本正常,除了雄性不育。小鼠中缺乏BCL-2会导致过度的BIM驱动的凋亡,减少成熟B细胞和T淋巴细胞的数量,并导致过早白发以及致命的多囊肾病。BCL-XL缺乏在小鼠发育的胚胎第13天是致命的,因为这妨碍了血小板以及某些神经和红细胞群体的存活。MCL-1的缺失影响最为显著,对于早期植入胚胎和广泛的细胞类型的存活至关重要,包括造血细胞群、心肌细胞和肠上皮细胞。
在某些细胞类型中,必须消除两种促生存BCL-2蛋白才能观察到严重缺陷。例如,BCL-XL或MCL-1可以维持肝细胞和某些神经细胞群的存活。促生存BCL-2蛋白和BH3-only protein的水平被精细平衡,因为基因剂量减半可能会产生戏剧性后果。例如,Mcl-1+/− Bcl-x+/−小鼠在出生后不久即死亡,表现出严重的颅面异常,但Mcl-1+/− Bcl-x+/− Bim+/−小鼠则健康。
由缺陷的内源性凋亡引起的疾病
如上所述,强制表达BCL-2抑制内源性凋亡,可能导致人类滤泡性淋巴瘤。在小鼠中,淋巴细胞转基因过度表达BCL-2会引起类似于系统性红斑狼疮(SLE, systemic lupus erythematosus)的致命自身免疫疾病和低发病率的淋巴瘤。这些发现突显了内源性凋亡在维护免疫耐受以及肿瘤生成中的重要性。值得注意的是,当造血细胞还有MYC的非调节表达时,BCL-2是一个更强大的致癌基因,MYC是一个推动异常细胞分裂的转录因子。过量的BCL-2使恶性和非转化细胞对广泛的抗癌药物产生抵抗,无论这些药物是以p53依赖还是非p53依赖的方式杀死细胞。特别是BIM和PUMA这样的BH3-only protein未能中和过量的BCL-2,这阻止了内源性凋亡。因此,为癌症治疗开发了模仿BH3-only protein功能的小分子BH3类似物。特定于BCL-2的BH3类似物Venetoclax(也称为ABT-199)目前已被许多监管机构批准用于治疗慢性淋巴细胞白血病(CLL, chronic lymphocytic leukemia)和急性髓性白血病(AML, acute myeloid leukemia),这些恶性病变主要依赖BCL-2而非其他促生存BCL-2蛋白来生存。针对MCL-1和BCL-XL的BH3类似物也已被开发,但它们在正常健康细胞中的靶向毒性构成了一个挑战。针对BCL-XL的治疗可能会导致血小板减少症,而心肌细胞、肠上皮细胞和血液细胞亚群是一些可能不耐受MCL-1抑制剂的细胞类型。将BH3类似物选择性地送达癌细胞的抗体偶联物的使用可能是限制正常健康细胞毒性的策略。
在某些情况下,抑制内源性凋亡可以预防而非促进肿瘤发展。例如,在小鼠中,低剂量γ-射线诱导的胸腺淋巴瘤可被Puma缺陷所预防。解释是,成熟白细胞应激诱导的凋亡触发了大量的造血祖细胞的动员和增殖,从而产生了肿瘤原性病变(oncogenic lesion)。MCL-1缺陷导致的增强凋亡促进了小鼠肠道的肿瘤生成。因此,了解BH3类似物药物诱导的正常细胞凋亡是否可能为次发性治疗相关癌症埋下伏笔,就显得很重要了。
过度的内源性凋亡与急性和慢性退行性疾病有关,包括缺血再灌注损伤和神经退行性疾病。至今为止,针对caspase的治疗策略并未取得成功,抑制剂在临床试验中展现出有限的疗效以及毒性。重要的是,抑制内源性凋亡途径中的caspase并不能阻止线粒体外膜通透性改变(MOMP)后的线粒体功能障碍和细胞死亡。目前正在努力寻找抑制BAX或BAK的抑制剂,但考虑到可能的靶向毒性,是否存在治疗窗口尚不清楚。
外源性凋亡和坏死性凋亡
外源性凋亡和坏死性凋亡途径主要由细胞表面的死亡受体结合的细胞外配体触发。通过caspase-8传递凋亡信号,而当caspase-8被抑制时,可能发生坏死性凋亡,这是一种溶解性的细胞死亡形式。坏死性凋亡可能作为一种抗病毒防御机制而进化,因为某些病毒,包括巨细胞病毒、单纯疱疹病毒、牛痘病毒和腺病毒,编码caspase-8的抑制剂以防止凋亡。与此观点一致的是,缺乏坏死性凋亡的小鼠比野生型(WT)小鼠更易感染牛痘病毒或缺乏坏死性凋亡抑制剂病毒诱导的RIPK3降解(vIRD, viral inducer of RIPK3 degradation)的变异牛痘病毒。与内源性凋亡途径中的caspase-9一样,caspase-8通过裂解并因此激活caspase-3和-7来执行凋亡程序。
配体和死亡受体(death receptor)配对包括FAS配体(FASL)与FAS、肿瘤坏死因子(TNF)或淋巴毒素-α与肿瘤坏死因子受体1(TNFR1)、TNF样细胞因子1A(TL1A)与死亡受体3(DR3)以及与TRAILR1或TRAILR2(小鼠只有一个TRAILR)相关的诱导凋亡配体(TRAIL)。尽管它们的名字,死亡受体并不专门传递细胞死亡信号。许多细胞对TNFR1的激活不是通过死亡而是通过激活核因子κB(NF-κB)和丝裂原激活蛋白激酶(MAPK)信号通路,促进促炎症和促生存基因的表达。TNFR1信号机制的扰动可能会促使细胞死亡的诱导,并在下文中进行讨论。
死亡受体之外,外源性凋亡或坏死程序(necroptosis)的机制也可以通过 Toll 样受体 3(Toll-like receptor 3, TLR3)、TLR4 或 Z-DNA 结合蛋白 1(Z-DNA binding protein 1, ZBP1)被激活。TLR3 通过内体(endosomal compartment)中的双链 RNA 被激活,而 TLR4 感应细菌的外细胞脂多糖(lipopolysaccharide, LPS)并随后转移到内体。ZBP1 是内部的 Z-形核酸传感器,这种核酸具有左旋的双链螺旋结构,由一些病毒和内源性逆转录元件产生。TLR3 或 TLR4 信号的典型输出是促炎和促存活的基因表达,但如 TNFR1 信号传导所见,某些细胞缺陷可能会使信号传导倾向于细胞死亡。因此,当细胞暴露于某些死亡配体或病原体相关分子模式(PAMPs, pathogen-associated molecular patterns)时,如果立即受到威胁,反映在关键信号组件的损害上,它们可以将其反应从促炎和促存活基因表达升级为外源性凋亡或坏死程序。
TNFR1
TNFR1在许多细胞类型上表达,并在病原体防御中发挥重要作用,但持续激活该受体可能助长慢性炎症。事实上,TNF抑制剂,它们阻断TNF-TNFR1和 TNF-TNFR2信号,广泛用于治疗自发炎性疾病,如炎症性肠病、类风湿性关节炎和强直性脊柱炎。与所有死亡受体一样,TNFR1有一个细胞质同型蛋白互作基序(cytoplasmic homotypic protein interaction motif),称为死亡结构域(death domain, DD)。在配体诱导的TNFR1寡聚化后,DD通过招募与死亡结构域相关的TNFRSF1A适配蛋白(TNFRSF1A associated via death domain, TRADD)和受体相互作用蛋白激酶1(receptor interacting protein kinase 1, RIPK1)启动所谓的复合体I的组装(图 2A)。TRADD 绑定到 TNF 受体相关因子2(TNF receptor associated factor 2, TRAF2),这是 cIAP1 和 cIAP2 泛素连接酶的适配蛋白。cIAP1 和 cIAP2 通过向 RIPK1 和复合体 I 的其他组件添加K63连接的多聚泛素链,帮助对接线性泛素链装配复合体(linear ubiquitin chain assembly complex, LUBAC)和TGF-β激活的激酶1(TGF-beta activated kinase 1, TAK1)。LUBAC然后向TNFR1、TRADD 和 RIPK1添加M1连接的多聚泛素链,这增强了招募典型IκB激酶(IKK)复合体的能力。复合体I中TAK1和IKK 的激活,参与了NF-κB和MAPK 信号通路,驱动促炎基因(例如Ccl2, Ccl3, Ccl5, Csf2, Cxcl1, Cxcl2, Il1b, Il6, Nos2 和 Tnf)和促存活基因(例如Bclx和Cflar,编码细胞FLICE抑制蛋白[cFLIP]的基因)的表达。
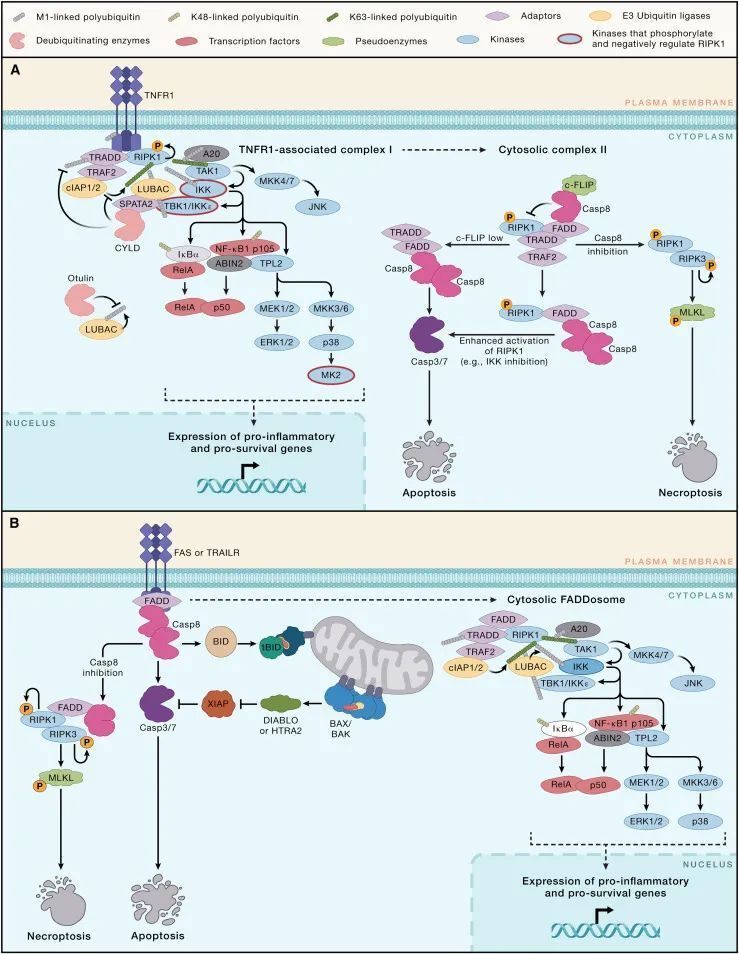
图 2-外源性凋亡信号通路(Credit: Cell)
线性泛素链装配复合体(LUBAC,linear ubiquitin chain assembly complex)也是 TANK 结合激酶 1(TBK1)和 IKKε 进入复合体 I 的必要条件。这些激酶通过磷酸化 RIPK1,从而抑制其诱导死亡的激酶活性。TNFR1 信号激活的其他激酶,包括 IKK 和 MK2(也称为 MAPK 激活的蛋白激酶 2),通过在 RIPK1 的不同位点磷酸化促进细胞存活。RIPK1 的 K63 连接泛素化也似乎抑制了其激酶活性。阻断 RIPK1 上的这些翻译后修饰会使 TNFR1 信号向细胞死亡倾斜。
TNFR1 下游 RIPK1 的激活如何促进细胞死亡仍在阐明中。RIPK1 的激酶活性对于 TNF 诱导的 NF-κB 或 MAPK 信号是非必需的,RIPK1 的主要底物似乎是其自身。RIPK1 在死亡结构域介导的激酶二聚化后的自磷酸化使 RIPK1 获得进入次级细胞质诱导死亡的复合体 II 的资格。RIPK1 从复合体 I 到复合体 II 的转变由圆柱状纤维瘤症(cylindromatosis, CYLD)增强,这是一种去泛素化酶,可以移除 M1 或 K63 连接的多聚泛素。CYLD 通过其适配蛋白: 精子生成相关蛋白 2(SPATA2)被招募到复合体 I,后者又与 LUBAC 结合。CYLD 是否能够接触其底物似乎还受到泛素结合蛋白 A20(也称为 TNF alpha 诱导的蛋白 3)和 A20 结合并抑制 NF-κB(ABIN-1)的进一步控制。这些蛋白都被招募进入复合体 I,并可能通过 CYLD 抑制多聚泛素链的切割。据此模型,缺乏 ABIN-1 和 A20 的小鼠肠上皮细胞比其 WT 对应物更易于 TNF 诱导的细胞死亡。
使用环己酰胺抑制蛋白质翻译也可以增强 TNF 诱导的细胞死亡敏感性,但在这种情况下,RIPK1 的激酶活性是非必需的。TRADD 可以独立于 RIPK1 成核复合体 II。无论是 TRADD 还是 RIPK1 形成复合体 II 的骨架,两种蛋白都可以与通过死亡结构域相关的 FAS(FADD)结合,这是 caspase-8 的适配蛋白。FADD 中的死亡效应结构域(DED)然后与 caspase-8 酶原体中的两个 DED 之一相互作用。caspase-8 中的串联 DED 支持由 caspase-8 及其催化不活跃的同源物 cFLIP 组成的螺旋状 DED 丝状体的组装。
caspase-8 的近邻诱导的同源二聚体,或者 caspase-8 与长型 cFLIP(cFLIPL)的异源二聚体,自我蛋白水解处理产生完全活跃的 caspase-8。caspase-8 的催化亚基之间的切割允许构象变化,稳定活性位点,但在 caspase-8/cFLIPL 异源二聚体中这一点不那么关键。异源二聚体介导的 caspase-8 同源二聚体的切割可能促进 caspase-8 的激活,至少在最初阶段如此。caspase-8 与 cFLIPL 的比例很重要,因为丰富的 cFLIPL 可以通过限制 DED 寡聚体中合并的 caspase-8 的总量来抑制 caspase-8 的激活。较短的 cFLIP 异构体(例如人类 cFLIPS 和病毒 FLIP 蛋白)通过扰乱定向 caspase-8 二聚体的螺旋状 DED 丝状体来抑制 caspase-8 的激活。环己酰胺似乎通过减少高度不稳定的 cFLIPL 量来促进 TNF 诱导的凋亡。cFLIPL 丰度的严格调控可能是为了促进快速杀死蛋白质翻译受损的病毒感染细胞而进化的。一些病毒获得了自己的 cFLIPS 版本,作为阻断外源性凋亡的手段。其他病原体,包括肠致病性大肠杆菌(EPEC),也获得了其他阻止死亡受体信号传导的手段。例如,EPEC 毒力因子 NleB1 是一种乙酰葡萄糖胺转移酶,可以修改并禁用 DD,包括 TNFR1、TRADD、RIPK1 和 FADD 中的那些。
许多未感染的原代细胞对 TNF 的反应是低水平的 caspase-8 激活,不会触发凋亡。现有证据表明,在这种情况下,caspase-8 切割复合体 II 中的 RIPK1 支架,从而在有足够活跃的 caspase-8 有效切割和激活 caspase-3 和 -7 之前破坏复合体 II。与此模型一致的是,消除 RIPK1 中 caspase-8 切割位点的杂合突变增强了 TNF 诱导的 caspase-8 激活和凋亡。XIAP,作为 caspase-3、-7 和 -9 的抑制剂,可能增加了一层保护,防止凋亡的诱导。报道的其他限制复合体 II 的机制包括通过自噬相关蛋白 9A (ATG9A) 将复合体靶向溶酶体,以及通过聚 ADP 核糖聚合酶 tankyrase-1 的多聚 ADP 核糖化和随后的泛素化而导致的复合体的蛋白酶体降解。
健康细胞中TNFR1对caspase-8的“挠痒”并非徒劳,因为它为当病原体抑制caspase-8时稳定化诱导死亡的复合体II(Complex II)奠定了基础。如果在复合体II中,活跃的RIPK1未被caspase-8切割,则它可以激活激酶RIPK3,释放caspase独立的混合谱系激酶结构域样(Mixed Lineage Kinase Domain-Like, MLKL)介导的坏死性死亡(necroptosis,如下所述)。因此,通过疱疹病毒B13R、牛痘病毒CrmA或小分子泛caspase抑制剂对caspase-8的抑制,会使某些细胞对TNF诱导的细胞死亡更加敏感。RIPK1和RIPK3通过它们的RIP同型互作基序(RIP Homotypic Interaction Motifs, RHIMs)相互作用。RHIM驱动的RIPK3寡聚化激活其激酶活性,导致伪激酶MLKL的磷酸化。MLKL随后形成寡聚体并转移到膜上,引发细胞裂解。坏死性死亡作为一种抗病毒防御机制的重要性,通过发现阻止RIPK3激活MLKL的病毒MLKL样假体(decoys)而凸显出来。已经鉴定出通过干扰RHIM依赖的RIPK3激活或针对RIPK3进行蛋白酶体降解来阻断坏死性死亡的病毒和细菌蛋白。
虽然人类RIPK3的功能丧失突变与单纯疱疹脑炎(herpes simplex encephalitis)有关,但目前尚不清楚该疾病是否源于病毒感染细胞坏死性死亡受损,因为RIPK3还有坏死性死亡独立的功能。与RIPK3坏死性死亡独立作用一致,极少数MLKL缺陷病例未表现出对传染性病原体的增加敏感性。
在小鼠中,肠上皮细胞或角质细胞中过量的TNFR1驱动细胞死亡是炎症的强劲驱动因素。例如,在LUBAC功能受损的Sharpincpdm突变小鼠中,慢性增生性皮肤炎可通过消除TNFR1、CYLD、FADD、caspase-8或RIPK1的激酶活性而被阻止。由于肠上皮细胞中IKK亚单位NEMO的丧失引起的结肠炎也可通过丢失TNFR1、FADD或RIPK1的激酶活性而被阻止。因此,即使在没有促炎症NF-κB信号的情况下,TNFR1诱导的细胞死亡也能驱动炎症。过度的凋亡可能会破坏皮肤或肠道这些屏障,并允许微生物的涌入,进而驱动炎症。
在人类中,TNFR1诱导的细胞死亡被认为是由TBK1或OTULIN缺陷引起的自身炎症综合征的重要驱动因素。OTU去泛素化酶具有线性连接特异性(OTULIN, OTU deubiquitinase with linear linkage specificity)通过从泛素连接酶去除M1连结的多聚泛素来保持LUBAC活性。缺乏TBK1或OTULIN的患者细胞在培养中表现出对TNF诱导死亡的高度敏感性,值得注意的是,这些患者接受TNF抑制治疗可以缓解疾病。与人类不同,小鼠中Tbk1或Otulin失活突变会导致胚胎死亡。Tbk1缺陷导致胎儿肝脏中由TNFR1和RIPK1激酶活性驱动的细胞死亡,而Otulin失活(或丢失核心LUBAC基因Hoil1或Hoip)引起的胚胎致死部分依赖于TNFR1和活跃的RIPK1。缺乏NEMO的人类中的一些炎症病变对TNF阻断有反应,鉴于IKK缺陷增强了TNFR1刺激的细胞死亡,它们也可能反映了异常的TNF诱导细胞死亡。
编码A20的TNFAIP3的半数不足可以引起自身炎症和自身免疫症,有些患者对TNF阻断有反应。虽然A20缺陷增加了对TNF诱导细胞死亡的敏感性,但完全缺乏A20的小鼠会发展出独立于TNF或TNFR1信号的严重早发炎症。后一观察表明,TNF诱导的细胞死亡只是A20负向调控的几个促炎过程之一。表达低活性A20且具有失活的锌指7(Zinc Finger 7, ZnF7)泛素结合结构域的小鼠会发展出TNF驱动的关节炎,而在A20中同时突变ZnF4和ZnF7会引发与A20缺陷相关的更严重表型。在完全缺乏A20或A20 ZnF4/ZnF7突变小鼠中消除依赖caspase-8的细胞死亡和依赖MLKL的坏死性死亡的影响尚未报道。因此,尚不清楚死亡受体或TRIF依赖的TLRs引发的异常细胞死亡是否是其致死性的主要驱动因素。
TNFR1(肿瘤坏死因子受体1)刺激细胞死亡在没有通路负调控因子基因突变失活的情况下,其在人类炎症性疾病中的作用尚不明确。尽管如下所述,RIPK1 (受体相互作用蛋白激酶1)抑制剂的临床试验可能提供一些见解,但是抑制RIPK1的激酶活性也可能阻止FAS、TRAILR1、TRAILR2、TLR3和TLR4触发的某些形式的细胞死亡。接受RIPK1抑制剂GSK2982772治疗的类风湿关节炎和结肠炎患者在临床试验中未显示出益处(ClinicalTrials.gov 研究: NCT02858492和NCT02903966),但其他RIPK1抑制剂正在皮肤红斑狼疮(ClinicalTrials.gov 研究: NCT04781816)和肌萎缩侧索硬化症(ClinicalTrials.gov 研究: NCT05237284)的临床试验中。
DR3
DR3的表达范围不如TNFR1广泛,主要发现于淋巴细胞上,但它似乎与TNFR1以相似方式发出信号。TL1A (肿瘤坏死因子样配体1A) 与DR3的结合主要刺激促炎细胞因子和趋化因子的产生,但如cIAP缺乏这类扰动会促进细胞质中诱导信号复合体的组装。TL1A抑制剂已被证明对炎症性肠病患者有益,这表明TL1A像TNF一样,是慢性炎症的重要驱动因子。
FAS和TRAIL受体
FAS、TRAILR1和TRAILR2这些死亡受体也通过两个连续的复合体发出信号,但顺序与TNFR1和DR3相反。与受体相关的复合体触发细胞死亡,而第二个细胞质复合体称为FADDosome(FADD相关复合体),在未能死亡的细胞中可以刺激NF-κB和MAPK信号通路。FAS或TRAIL受体的DD(死亡结构域)直接与FADD结合以激活caspase-8。尽管caspase-8可以直接切割并激活caspase-3和-7,但FAS在“第二型”细胞(例如小鼠肝细胞)中诱导的凋亡需要caspase-8介导的促凋亡BH3仅限蛋白BID的蛋白水解激活,产生tBID。需要激活内在途径以克服XIAP介导的caspase-3和-7的抑制。FAS和TRAIL受体像TNFR1一样,在caspase-8被抑制时可以信号触发坏死性凋亡,这种细胞死亡依赖于RIPK1的激酶活性。RIPK1被FADD招募至受体复合体,随后与RIPK3结合促进坏死性凋亡。
FADDosome的组装需要FADD和caspase-8,但不需要caspase-8的催化活性。FADDosome似乎包含了TNFR1复合体I的许多组分,包括TRADD、cIAP1/2、RIPK1、TRAF2、IKK、TAK1和A20。对于抵抗TRAIL诱导细胞死亡的肿瘤细胞,FADDosome调控的基因表达已被提出可调节抗肿瘤免疫细胞反应。
FASL和TNF是细胞毒性T细胞用来杀死其靶细胞的武器,除了穿孔素和颗粒酶。过度的TNFR1驱动细胞死亡和过度的FAS或TRAILR触发的细胞死亡都可能导致组织损伤和炎症。FASL还通过杀死不再需要的淋巴细胞来维持淋巴稳态。FASL或FAS功能受损的小鼠和人类会发展成自身免疫性淋巴增生综合征(ALPS),一种通常稀有的、非传统B和T细胞异常累积的疾病。FADD或caspase-8酶活性的缺失(阻止所有外源性凋亡)对小鼠和人类的影响不同。无法激活caspase-8的小鼠会因未受控的TNFR1-、RIPK1-、RIPK3-和MLKL依赖的坏死性凋亡而在胚胎期死亡。相比之下,缺乏FADD或caspase-8的人类可能表现出ALPS、反复感染和早发性炎症性肠病。是否坏死性凋亡驱动了人类疾病尚不清楚。受损的caspase-8切割RIPK1必须促进炎症,因为杂合性RIPK1突变破坏了caspase-8切割位点,导致周期性发热综合征。NEDD4结合蛋白1(N4BP1)的切割和失活可能导致FADD或caspase-8缺陷患者的先天免疫受损。在人和小鼠的骨髓细胞中,N4BP1通过仅通过髓样分化基因88(MyD88)适配器信号的TLRs抑制细胞因子和趋化因子的产生。N4BP1是一种泛素结合内核糖核酸酶,但它如何精确地限制细胞因子和趋化因子反应还需进一步研究。
TLR3、TLR4和ZBP1
TLR3和TLR4都利用含RHIM(RIP同源相互作用基序)的适配蛋白Toll/IL-1受体域含有诱导干扰素-β的适配蛋白(TRIF)来与RIPK1和RIPK3结合(图3A)。反过来,RIPK1可能招募TRADD-TRAF2-cIAP1/2和FADD-caspase-8,最终导致NF-κB和MAPK激活以及caspase-8依赖的N4BP1切割。已显示阻断LUBAC或同时阻断cIAP1和cIAP2可使TLR3信号转向强烈的caspase-8激活和细胞死亡。如果caspase-8被抑制,那么TLR3和TLR4都可以刺激坏死性凋亡。抑制RIPK1可阻止巨噬细胞中TLR3或TLR4诱导的坏死性凋亡,这表明RIPK1的激酶活性通常是TRIF依赖性坏死性凋亡所必需的。然而,小鼠的遗传研究表明,即使在RIPK1缺失的情况下,TRIF或ZBP1也可以激活坏死性凋亡。实际上,小鼠RIPK1似乎利用其RHIM来抑制TRIF或ZBP1诱导的坏死性凋亡,因为RIPK1 RHIM突变小鼠中的炎症和围产期死亡可以通过失去MLKL或同时失去TRIF和ZBP1来预防。与RIPK1阻止TRIF从事细胞死亡机械相一致,缺乏RIPK1的原代人成纤维细胞对LPS或TLR3激动剂poly(I:C)诱导的细胞死亡更为敏感。总体而言,这些数据表明RIPK1的激活可能缓解其RHIM依赖性但又不甚明了的TRIF或ZBP1信号抑制。
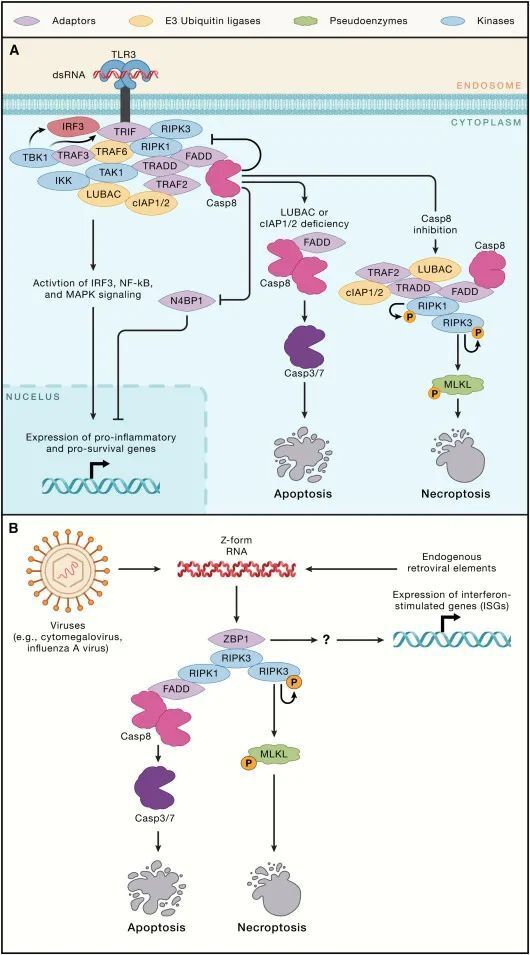
图3-TLR3和ZBP1的细胞死亡信号(Credit: Cell)
RIPK1支架也抑制caspase-8驱动的细胞死亡,因为Ripk1缺失的小鼠在围产期死亡,除非消除caspase-8和MLKL依赖的死亡程序。相比之下,RIPK1的激酶活性对小鼠的存活来说是可有可无的。RIPK1似乎至少部分限制凋亡,是通过阻止TRADD-FADD-caspase-8信号复合体的组装。考虑到RIPK1和TRADD似乎并行作用并且绑定相同的DD蛋白,RIPK1的缺失可能允许更多TRADD进入信号复合体,同时伴随着NF-κB和MAPK信号受损和cFLIPL表达减少。RIPK1缺失的原代人成纤维细胞也增强了TNF诱导的细胞死亡。尽管RIPK1的缺失在小鼠中是致命的,但缺乏RIPK1的人类主要表现为免疫细胞功能障碍,导致淋巴细胞减少、感染、关节炎和早发性炎症性肠病。小鼠与人类对RIPK1的更广泛需求可能反映了物种特异性差异,例如RIPK1的表达模式或其他通路组分的差异。
ZBP1具有两个Zα结构域用于识别Z型核酸和一个RHIM与RIPK3结合。ZBP1-RIPK3相互作用可以通过MLKL触发坏死性凋亡或通过RIPK1、FADD和caspase-8触发凋亡(图3B)。RIPK1的激酶活性对这种细胞死亡来说是非必需的。制造Z-RNA的病毒包括巨细胞病毒、流感A病毒、单纯疱疹病毒1和牛痘病毒。巨细胞病毒通过产生caspase-8和RHIM-RHIM相互作用的抑制剂来逃避细胞死亡,而牛痘病毒则产生caspase抑制剂和一个Zα结构域蛋白,用以屏蔽病毒Z-RNA免受ZBP1的检测。内源性逆转录病毒元件产生的Z型核酸在过度的ZBP1依赖性细胞死亡导致皮肤或肠道炎症的小鼠模型中被暗示。
有趣的是,ZBP1信号的输出不仅限于细胞死亡。ZBP1还可以独立于RIPK3、RIPK1 RHIM或细胞死亡,促进干扰素刺激基因(ISGs)的表达。解开这一信号通路将是重要的,因为ZBP1对由Adar1突变导致的ISG驱动病理有所贡献。人类ADAR1突变引起的重症特征是ISG表达增加,包括艾卡蒂-古蒂耶症候群(Aicardi-Goutieres syndrome)。特异性腺苷脱氨酶RNA(ADAR1)具有Zα结构域,似乎绑定并编辑会否则激活ZBP1的Z-RNAs。鉴于ZBP1诱导的坏死性凋亡和ISG表达的促炎性质,其他人提出利用ZBP1激动剂来增强癌症免疫疗法。
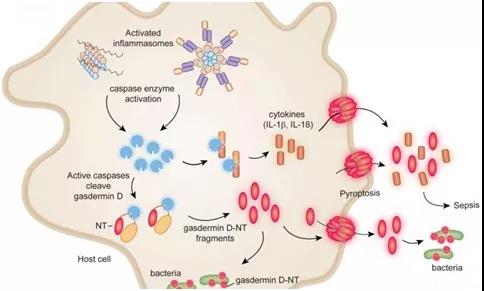
焦亡(Pyroptosis)
焦亡是指由细胞质膜上的gasdermin孔洞引起的细胞死亡。焦亡作为一种独特的细胞死亡程序,起源于对感染沙门氏菌的巨噬细胞的研究。这种感染巨噬细胞的死亡依赖于caspase-1,这是一种蛋白酶,可以蛋白水解成熟IL-18和内源性发热原IL-1β。后续研究揭示,在人类中,caspase-1、-4和-5,以及在小鼠中,caspase-1和-11,都可以通过切割gasdermin D(GSDMD)引起焦亡。GSDMD的N端切割片段通过寡聚化并在膜上形成孔洞,促进细胞裂解。GSDMD在细胞膜上的孔洞会破坏离子梯度,并导致水分的大量涌入,最终引发依赖于ninjurin 1(NINJ1)的细胞质膜破裂(PMR)以及大量促炎DAMPs的释放。然而,包括IL-1β在内的小型DAMPs可以通过GSDMD孔洞离开细胞。在某些情况下,细胞膜修复机制可能限制GSDMD孔洞的形成。GSDMD孔洞是否能在不触发焦亡的情况下释放IL-1β仍未解决。
炎症小体(Inflammasomes)
Caspase-1在细胞质复合体中形成活性二聚体,这些复合体被称为炎症小体(图4)。炎症小体的组装由感应特定DAMPs或PAMPs的蛋白质触发,然后形成寡聚骨架。一些“感应器”蛋白质直接与DAMPs或PAMPs结合,而其他蛋白质则检测DAMPs或PAMPs在细胞内引起的扰动。前者包括absent in melanoma 2(AIM2),它与病毒或细菌的双链DNA结合,以及NLR家族凋亡抑制蛋白(NAIPs),它们与细菌的鞭毛、针和内杆蛋白结合。NLR家族含有pyrin结构域的1(NLRP1)和caspase激活和招募结构域8(CARD8)能够感知某些感染,因为它们各自具有一个抑制结构域,可以被细菌或病毒酶修饰并靶向蛋白酶体降解。这种抑制结构域的去除促进了炎症小体的组装。
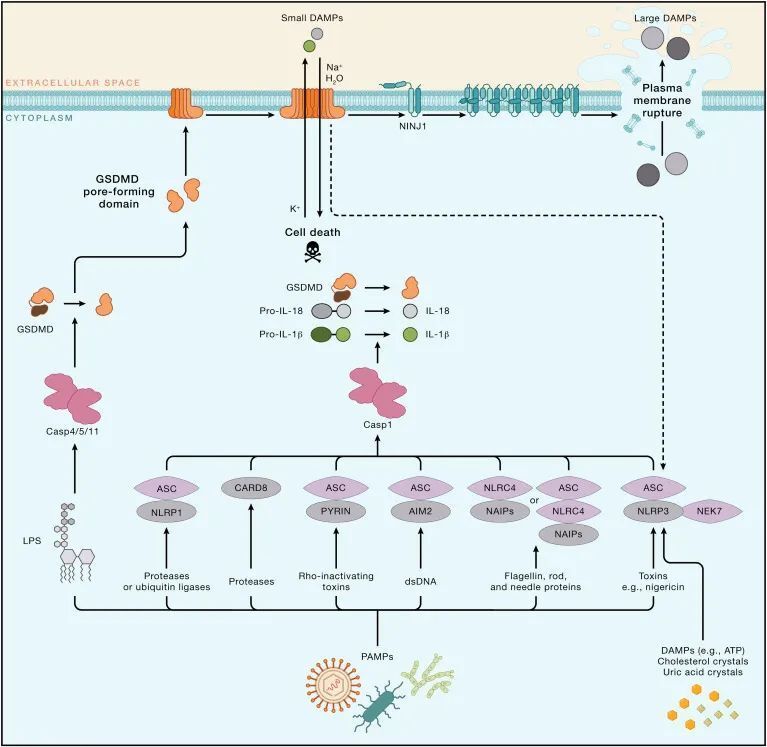
图4-炎症小体诱导的GSDMD依赖性焦亡(Credit: Cell)
识别PAMPs或DAMPs的炎症小体感应器包括PYRIN和NLRP3。PYRIN能够感知修改和失活Rho GTP酶的细菌毒素,PYRIN的去磷酸化和微管聚合在炎症小体组装中发挥作用。NLRP3对多种刺激物作出反应,包括细菌毒素nigericin、胞外ATP和与痛风相关的尿酸晶体。细胞内钾离子流出被认为是驱动NEK7依赖性NLRP3寡聚化的统一事件,但这一过程的机制细节仍然不清楚。NLRP3在无菌炎症的背景下激活促使了小分子NLRP3抑制剂的开发。NLRP3抑制剂的临床试验集中在自身炎症性疾病上,包括骨关节炎、神经炎症以及COVID-19。PYRIN、NLRP1和NLRP3-NIMA相关激酶7(NEK7)炎症小体都包含了caspase-1适配蛋白含有CARD的凋亡相关斑点状蛋白(ASC),它有一个CARD与caspase-1中的CARD结合。NAIP和CARD8炎症小体不需要ASC,因为NAIP相互作用蛋白NLR家族含有CARD的4(NLRC4)中的CARD或CARD8本身可以与caspase-1结合。
caspase-4或-5(啮齿动物caspase-11的人类对应物)中的CARD直接与细菌LPS的细胞质寡聚体结合。这种结合不需要表面表达的LPS受体TLR4。这些非典型炎症小体复合物产生活性的caspase二聚体,触发GSDMD依赖性焦亡。焦亡细胞还释放成熟的IL-1β,因为GSDMD切割引起的扰动激活NLRP3炎症小体和caspase-1。
GSDMD依赖性焦亡在病原体防御和炎症性疾病中的作用
小鼠的遗传研究表明,GSDMD依赖性焦亡是限制沙门氏菌等细胞内病原体生长的重要机制。其他细胞内细菌通过使用抑制焦亡的毒力因子来保护其复制生态位。志贺氏菌有两个效应器武器来阻止焦亡,caspase-4和-11的抑制剂OspC3和泛素连接酶IpaH7.8。后者通过将人类GSDMD和GSDMB(下文将描述)靶向蛋白酶体降解来抑制焦亡。
焦亡的促炎性质有助于抗击病原体,因为包括中性粒细胞在内的免疫细胞被招募到感染部位。但是,由于通路失调导致的过度焦亡会引起疾病。人类NLRP1、NLRP3、NLRC4和MEFV(编码PYRIN)的生殖系激活突变破坏了正常的炎症小体调节,并引起一系列自身炎症性疾病。这些疾病的患者通常对IL-1β阻断反应良好,异常的IL-1β产生可能反映了焦亡的增强。与这一观点一致的是,Gsdmd的缺失阻止了表达疾病相关PYRIN或NLRP3突变体的基因敲入小鼠中炎症性病变和活性IL-1β水平的升高。值得注意的是,在体外培养细胞中,Gsdmd缺失延缓而不是阻止了经典炎症小体激活后的细胞死亡和IL-1β释放。细胞死亡仍然发生,因为caspase-1还可以通过蛋白水解激活caspase-3和-7来触发较慢的凋亡过程。因此,体内的细胞死亡类型很重要。可以推测,在Mefv或Nlrp3基因敲入突变小鼠中,Gsdmd缺陷细胞通过凋亡死亡可能有益,因为在继发膜损伤释放由caspase-1产生的促炎、成熟形式的IL-1β之前,它们被吞噬细胞清除。
Gsdmd缺失还改善了小鼠败血症模型中的疾病。caspase-1、IL-1β和IL-18对小鼠LPS诱导的败血性休克是可有可无的,而caspase-11依赖性焦亡在内皮细胞中似乎驱动血管泄漏和低血压。通过内皮GSDMD孔洞的Ca2+流入可能促进微血管中致命的凝血和血块形成。总体来看,这些研究表明GSDMD抑制剂在暴发性炎症环境中可能提供治疗益处。有趣的是,最近的研究报告称,外源性添加的拮抗性GSDMD纳米体能够抑制体外细胞中的GSDMD依赖性焦亡。据称,GSDMD纳米体通过最初的GSDMD孔洞进入细胞,然后抑制进一步孔洞的形成。最初的GSDMD孔洞被认为是短暂的,并且通过膜修复机制从质膜中移除。这些发现表明GSDMD是可药用的,但这些纳米体是否能在体内抑制GSDMD尚不清楚。由于纳米体只能结合人类GSDMD,因此无法在临床前小鼠模型中评估其疗效。
除了半caspase-1、-4、-5和-11,还有其他蛋白酶也能切割并激活GSDMD(Gasdermin D)。由caspase-8切割GSDMD引起的细胞焦亡在应对耶尔森菌感染的宿主反应中扮演了重要角色。中性粒细胞弹力酶也被证实能够切割并激活GSDMD。相比之下,据报道caspase-3通过在N端形成孔的域内切割来使GSDMD失活。
更广泛的气道上皮细胞死亡蛋白家族
人类有六种气道上皮细胞死亡蛋白家族成员(GSDMA, GSDMB, GSDMC, GSDMD, GSDME, 和 GSDMF),而小鼠缺少Gsdmb,但有三种Gsdma基因(Gsdma1, Gsdma2, 和 Gsdma3),四种Gsdmc基因(Gsdmc1, Gsdmc2, Gsdmc3, 和 Gsdmc4),以及Gsdmd, Gsdme, 和 Gsdmf。除了GSDMF,每种气道上皮细胞死亡蛋白都有一个在N端的保守形成孔的域,被C端控制着。不同的蛋白酶切割不同的气道上皮细胞死亡蛋白以释放形成孔的域,这些域随后可以与细胞膜、细胞器膜或细菌的外膜结合。缺乏Gsdma1(或Gsdma1-3)、Gsdmc1-4、或Gsdme的小鼠比野生型对照组对某些细菌或蠕虫感染更易感,这表明许多气道上皮细胞死亡蛋白有助于宿主对病原体的防御。
在培养细胞中,caspase-3对GSDME的蛋白水解激活被报告可以引起细胞焦亡或凋亡后的次级膜损伤。后者的生理意义不太确定,因为在体内吞噬细胞通常会迅速清除凋亡细胞。无论如何,GSDME已被证实在小鼠中介导化疗药物引起的组织损伤或感染肠道病毒71型。
由细胞毒性淋巴细胞释放的Granzyme B可以切割并激活caspase-3来切割GSDME,但它也可以直接在靶向肿瘤细胞中切割GSDME。在各种肿瘤细胞中,GSDME的表观遗传沉默或突变失活暗示了GSDME在抑制肿瘤中的作用。因此,进行实验以查看Gsdme缺陷是否增强小鼠自发性肿瘤模型中的肿瘤发生将是有启发性的。已有报告称,嵌合抗原受体T细胞引起的GSDME驱动的肿瘤细胞裂解在小鼠中触发细胞因子释放综合征。
GSDMA主要在上皮细胞中表达。它似乎直接作为病原体传感器,通过链球菌致热外毒素B(SpeB)的蛋白水解切割后诱导细胞焦亡。GSDMB可以被细胞毒性淋巴细胞释放的Granzyme A切割并激活,Granzyme A是另一种效应蛋白酶。值得注意的是,已在不同细胞类型中描述了多种GSDMB剪接变体,其中一些编码无功能的GSDMB蛋白,这些蛋白不能诱导细胞焦亡。哪些亚型最具生理相关性尚不清楚,这使得解释将GSDMB的单核苷酸多态性与哮喘、克罗恩病和溃疡性结肠炎相关联的全基因组关联研究变得复杂。较少研究的GSDMC可能通过从杯状细胞和潘氏细胞释放IL-33在小鼠中介导抗蠕虫防御。
NINJ1依赖的质膜修复(PMR)
由NINJ1介导的PMR负责焦亡细胞的破裂,NINJ1是一种表达在细胞表面的跨膜(TM)蛋白。消除NINJ1并不会阻止细胞死亡,但会阻碍大型细胞内分子(如乳酸脱氢酶(LDH),PMR的标准标记物,和HMGB1)的释放。在培养中诱导焦亡的Ninj1缺陷细胞因其持续膨胀的形态而显著。认为从死亡细胞中排出的大型DAMPs(损伤相关分子模式)可“警告”免疫细胞,因为缺乏Ninj1的小鼠比野生型对照组更易感染肠杆菌属鼠伤寒杆菌或伪结核耶尔森菌。
在活细胞中,NINJ1似乎是单体或二聚体,但在焦亡细胞中则是寡聚体。纯化的NINJ1形成的纤维在一个表面上是疏水的,而另一个表面上是亲水的,这表明NINJ1通过封顶膜边缘245或形成类似纳米盘的环,从而撕裂膜的部分。目前尚不清楚什么触发了垂死细胞中NINJ1的寡聚化。一种可能性是NINJ1感知到随着细胞膨胀而发生的膜张力或脂质排列的变化。
在培养中,NINJ1也介导凋亡细胞的次级PMR。凋亡细胞最终会膨胀,因为ATP水平下降和Na+/K+ ATPase泵功能失常。caspase-3对GSDME的切割和GSDME孔的形成可能会通过渗透压加速膨胀。有趣的是,NINJ1对经历坏死或铁死亡的细胞的PMR是可有可无的。NINJ1依赖的DAMP释放是否加剧炎症和疾病严重程度,是一个活跃的研究领域。
细胞死亡的灵活途径
有越来越多的证据表明哺乳动物细胞在编程性细胞死亡途径中的灵活使用。例如,巨噬细胞中的炎症小体可以激活caspase-1或-8,焦亡通常因为其更快的动力学而占优势。在小鼠中的遗传实验揭示了当通常占主导地位的死亡途径受阻时,细胞死亡的意外途径。例如,在小鼠中表达蛋白水解失活的caspase-8会激发致命的坏死,但如果消除MLKL以防止坏死,那么ASC和caspase-1信号传导则有助于致死性。在另一个例子中,FADD缺陷的小鼠肠上皮细胞的回肠炎是由坏死或GSDMD依赖的焦亡驱动的。最后,坏死可以触发细胞内NLRP3炎症小体的激活,导致caspase-1的激活和活化IL-1β的释放。假设NLRP3在垂死细胞内检测到某种形式的细胞扰动。尽管NLRP3和caspase-1不是这些细胞死亡所必需的,但它们仍然通过允许垂死细胞释放成熟的IL-1β,从而为死亡的炎症性质做出贡献。
铁死亡
铁死亡指的是细胞积累致命水平的依赖铁的磷脂过氧化物,从而导致非凋亡性死亡。经历铁死亡的细胞会破裂,但磷脂过氧化物如何破坏质膜的完整性尚不清楚。合成易氧化膜脂的代谢酶(例如,长链酰基辅酶A合成酶家族成员4 [ASCL4] 和溶血磷脂酰胆碱酰基转移酶3 [LPCAT3])和氧化膜脂的铁依赖酶(例如,花生四烯酸脂氧酶)赋予细胞对铁死亡的易感性(图5)。常用的铁死亡诱导剂包括小分子erastin和RSL3,它们通过干扰限制膜脂过氧化物的机制发挥作用。RSL3抑制谷胱甘肽过氧化物酶4(GPX4),这是一种利用谷胱甘肽将脂质过氧化物还原为无毒的脂质醇的脂质过氧化物酶。Erastin通过抑制进口半胱氨酸和使半胱氨酸可用于谷胱甘肽合成的xc−系统来耗尽细胞内谷胱甘肽。抑制其他自由基清除代谢物的产生,包括辅酶Q10(CoQ10)和四氢生物蝶呤(BH4),也可以使细胞对铁死亡更加敏感。增加游离铁的丰度是使细胞准备好铁死亡的另一种方式。
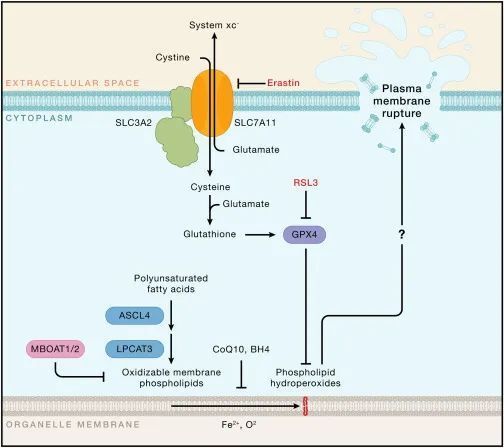
图5-细胞组分决定对铁死亡的敏感性(Credit: Cell)
结论和未来方向
细胞死亡对于多细胞生物的存活和适应性是必需的,但必须严格调节以防止疾病。对细胞死亡信号机制的全面理解揭示了在疾病中抵制异常细胞死亡的机会。就内在凋亡途径而言,成功的故事是BCL-2抑制剂Venetoclax的开发,用于治疗某些血液系统恶性疾病。基于在临床前炎症性疾病模型中遗传消除这些途径的益处,抑制促炎细胞死亡程序的抑制剂的识别是一项较新的努力。焦亡的效应器,如NLRP3和GSDMD,以及作为外源性凋亡和坏死途径的介质的RIPK1,是感兴趣的靶点。这些努力的关键是知道不同细胞死亡途径在人类疾病中何时何地被激活。各种形式的细胞死亡的分子标记已被定义,例如,坏死途径的磷酸化RIPK3或磷酸化MLKL,以及炎症小体诱导的焦亡的切割GSDMD,但它们在组织中的存在可能是短暂的,因为噬菌细胞会迅速清除死亡细胞。因此,即使在遗传学告诉我们途径处于活跃状态的临床前模型中,检测途径激活标记也可能具有挑战性。
原始出处:
https://doi.org/10.1016/j.cell.2023.11.044
 Cell综述:细胞死亡
Cell综述:细胞死亡
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#细胞死亡#
23