Genome Biology:王岱峰团队发表跨模态最优传输方法CMOT,可准确预测单细胞的多模态特征
2023-07-14 测序中国 测序中国 发表于上海

研究发现,在脑发育、癌症及免疫学等各种应用中,CMOT比现有方法更优秀,并提供改进细胞分类或癌症分类的生物学解释。
单细胞测序技术,例如scRNA-seq和scATAC-seq,可以提供单个细胞的多模态信息,包括基因组学、转录组学、表观基因组学、蛋白质组学等。多模态的单细胞测序技术检测为特定细胞和分子机制提供了全面理解。但与单模态检测相比,单细胞的多模态分析仍然具有挑战性,数据整合因缺失模态和细胞间对应而难以实现。
近日,美国威斯康星大学麦迪逊分校的王岱峰团队在Genome Biology上发表了题为“CMOT: Cross Modality Optimal Transport for multimodal inference”的研究文章。团队提出了跨模态最优传输(CMOT)方法,这是一种基于最优传输(OT)来推断单细胞缺失模式的计算方法,它将可用的多模态数据(源数据)中的细胞比对到一个共同的潜在空间,并推断另一模态(目标数据)中细胞的缺失模态,此推断并不需要成对的源数据多模态数据来进行比对。研究发现,在脑发育、癌症及免疫学等各种应用中,CMOT比现有方法更优秀,并提供改进细胞分类或癌症分类的生物学解释。

文章发表于Genome Biology
据文章介绍,CMOT是一种计算单细胞跨模态推理的计算方法,有三个主要步骤:步骤A-流形对齐,使源单元与多模式数据对齐。将具有可用的多模态数据细胞(源细胞)映射到一个共同的低维潜在空间,不同模态的细胞可以不配对或部分对应;步骤B-最优传输,通过共享的方式映射源细胞和目标细胞的最优传输。将具有单一形态的细胞(目标细胞)映射到来自同一形态的比对的源细胞;步骤C-K-近邻推理,K-近邻来推断目标细胞的额外形态。使用最近的映射源细胞的另一种模式来推断目标细胞的缺失或未分析的模式。(图1)
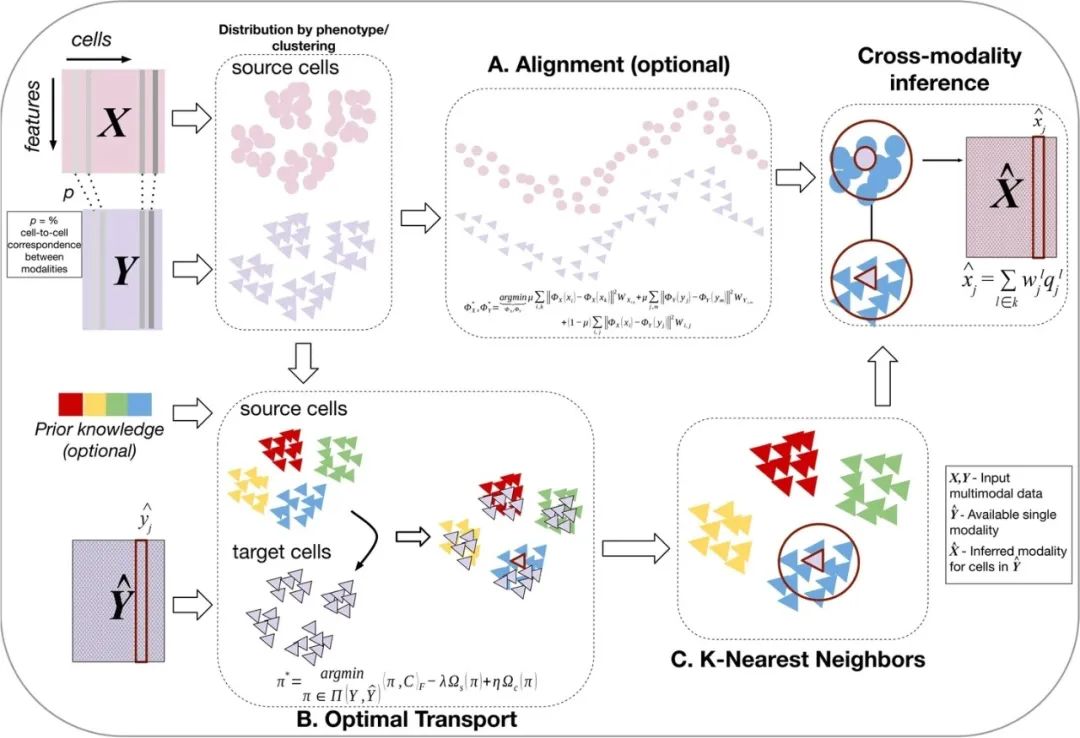
图1.跨模态最优传输(CMOT)的三个主要步骤。来源:Genome Biology
研究人员在4个单细胞多组学数据集上进行了CMOT评估:
(1)人类和小鼠大脑中单细胞的基因表达和染色质可及性(scRNA-seq&scATAC-seq);
(2)外周血单个核细胞的基因和蛋白表达(CITE-seq);
(3)A549肺癌细胞的基因表达和染色质可及性(sci-CAR);
(4) 胰腺癌细胞的基因表达和染色质可及性(scCAT-seq)。为了评估CMOT所推断的基因和蛋白表达,研究人员计算了每个细胞的推断表达值和检测表达值之间的皮尔逊相关系数,并且使用AUROC来评估CMOT推断开放染色质区域的峰的质量。
首先,研究人员使用CMOT从人和小鼠大脑染色质可及性推断单细胞基因表达。通过10xMultiome(scRNA-seq&scATAC-seq)联合分析基因表达和染色质可及性,并从开放染色质区域(scATAC-seq的峰值)使用CMOT推断细胞的基因表达。
结果表明,CMOT在基因表达推断方面表现优秀,优于Seurat和MOFA+等现有方法。根据CMOT推断基因表达区分的细胞类型获得了更高的AUROC,表明CMOT推断的基因表达能够区分细胞类型,CMOT推断单个细胞的表达具有较高的皮尔逊相关性和显著性。此外,研究人员从前100个高度预测的基因中发现了与大脑发育相关的丰富功能和通路。(图2)
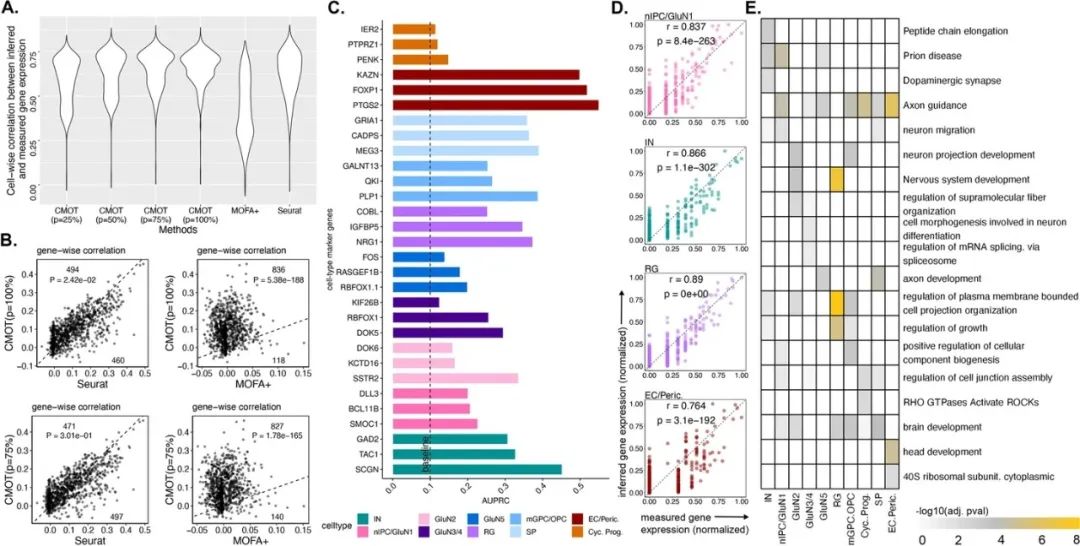
图2.从染色质可及性推断人脑单细胞基因表达。来源:Genome Biology
随后,研究人员应用CMOT从外周血单核细胞(PBMC)的基因表达中推断蛋白表达。研究人员使用来自CITE-seq数据PBMC10k的6885个细胞训练CMOT,然后使用来自不同数据集PBMC5k的3994个细胞评估了CMOT、MOFA+、Seurat和TotalVI。结果显示,CMOT推断和检测单个蛋白表达的皮尔逊相关性在所有情况下都相对较高,表明CMOT显著优于其他方法。(图3)

图3.从外周血单核细胞的基因表达推断蛋白质表达。来源:Genome Biology
接下来,研究人员评估了CMOT从染色质可及性推断经药物治疗的肺癌细胞基因表达的能力。将CMOT应用于100nM地塞米松(DEX)处理的肺腺癌A549单细胞,在处理0h、1h、3h后,使用sci-CAR实验对细胞的基因表达和开放染色质区域(OCR)进行分析,评估CMOT从OCR峰值信号推断基因表达方面的性能。结果显示,CMOT推断与检测的基因表达具有较高的皮尔逊相关性,表明CMOT在推断癌症细胞基因表达方面的能力优秀。(图4)
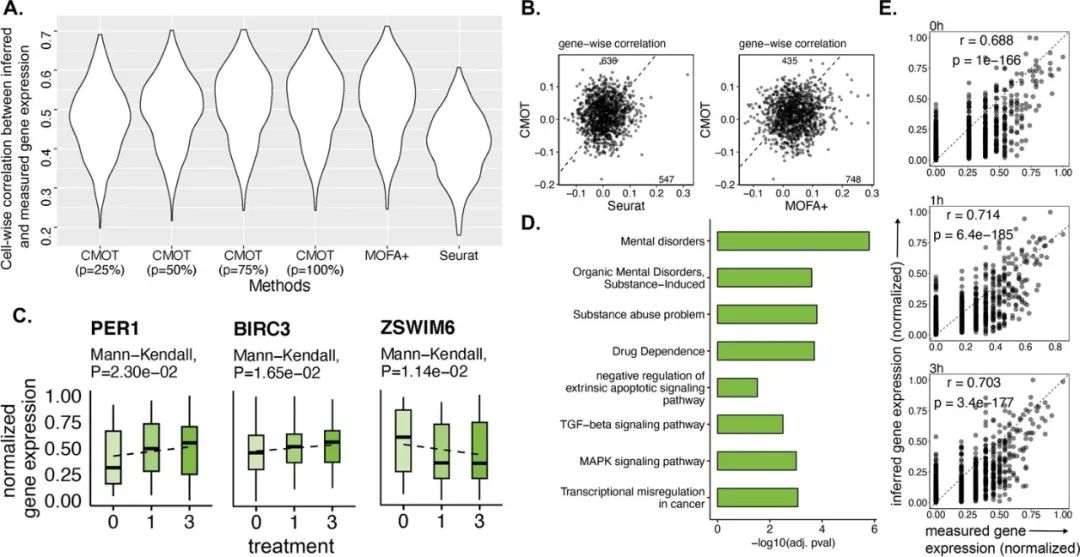
图4.利用染色质可及性推断药物治疗的癌症细胞的基因表达。来源:Genome Biology
最后,研究人员利用CMOT通过基因表达和染色质可及性之间的交叉模式推断来区分癌症类型。研究人员使用了一个泛癌scCAT-seq数据集训练CMOT,该数据集整合了三种癌症细胞系:HCT116、HeLa-S3和K562的OCR上的单细胞基因表达和染色质可及性。通过CMOT推断的基因表达,可以将三种癌细胞系准确分类,这表明CMOT推断能够揭示癌症类型特异性的表达。此外,在从基因表达推断OCR峰值方面,CMOT显著优于其他方法。(图5)
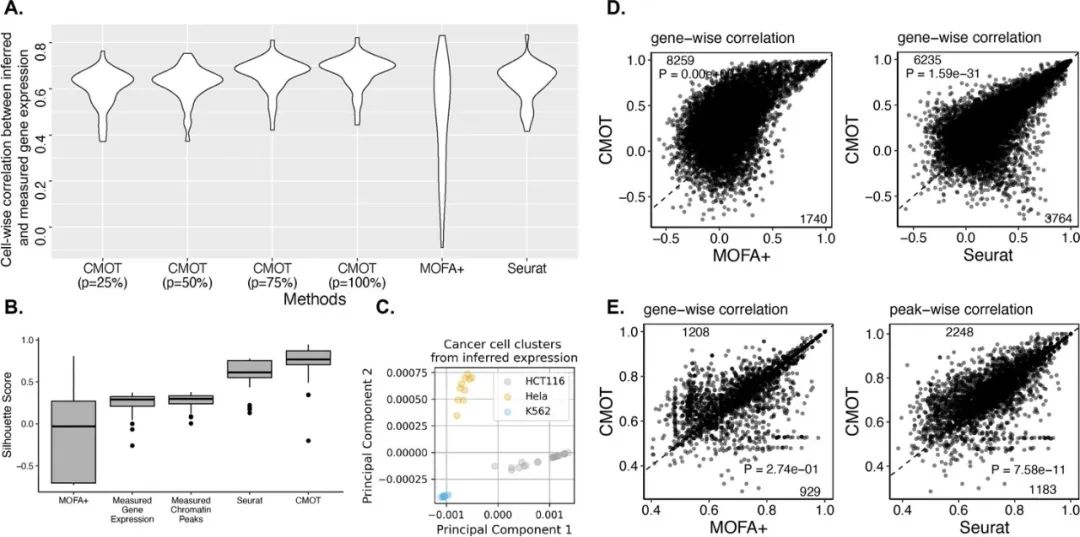
图5.基因表达和染色质可及性之间的交叉模式推断可以区分癌症类型 来源:Genome Biology
综上所述,CMOT是一种集成流形比对、最优传输和K-近邻推理用于交叉模态推理的计算方法。通过应用单细胞多模态数据,研究人员证明了CMOT能够预测单细胞的多模态特征,例如基因表达、染色质可及性和蛋白表达。CMOT不仅优于现有的先进方法,而且其推断的基因表达在生物学上可以解释,包括区分细胞类型和癌症类型。重要的是,CMOT不需要成对的样本来比对多种模式,克服了数据整合因缺失模态和细胞间对应的难题,领先于现有的其他方法。此外,CMOT的比对也具有灵活性,用户可以用其首选的比对方法来替代非线性流形比对。
原文链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-023-02989-8
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言