
Commun Biol:All of Us项目超9.8万名参与者的致病性变异频率分析揭示祖先驱动的差异
2024-03-01 测序中国 测序中国 发表于上海

该研究有助于针对性地开展精准医学工作,弥补医学研究中存在的不平等现象。
导读
实施基因组医学需要在真实世界的临床人群中注释基因组变异,但目前的大型基因组学研究队列通常缺乏多样性。据预测,普通人群中存在的大部分致病变异都是祖先群体所特有的。因此,要克服诊断率上的这一差距,就必须收集与电子健康记录数据相匹配的各种基因组数据。
All of Us研究旨在招募100万代表美国多样性的志愿者,生成一个独特的数据集,包含来自多样化参与者人群的基因数据、电子健康记录和身体测量数据,其中50.4%参与者以欧洲血统为主,23.2%以非洲血统为主,16.1%为拉丁裔/混合美洲血统等。该数据集可通过云计算平台(Researcher Workbench)进行共享,云计算平台包含98,590名参与者的全基因组测序(WGS)数据。
近日,All of Us项目研究团队在Communications Biology期刊发表了题为“The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities”的文章,系统分析了在All of Us队列中发现的致病性和可能的致病性变异。结果显示,不同祖先群体之间的致病性变异频率存在差异。欧洲血统亚组显示出最高的总体致病性变异率(2.26%);其他血统组的致病性变异率较低,非洲血统亚组为1.62%,拉丁裔/混合美洲血统亚组为1.32%。此外,致病性变异最常见于与乳腺癌/卵巢癌或高胆固醇血症相关的基因。该研究有助于针对性地开展精准医学工作,弥补医学研究中存在的不平等现象。

文章发表在Communications Biology
主要研究内容
All of Us数据集的致病性变异率
为了解All of Us数据中按预测祖先群体细分的已知致病性变异的比例,研究团队使用“VIP”数据库对项目参与者WGS数据中存在的P/LP变异进行注释,这些变异涉及73个具有可操作次要发现的基因。结果表明,欧洲血统组的已知致病变异率最高,其次是“其他”血统组和非洲血统组;致病变异率在非洲血统、欧洲和混合美洲/拉丁裔人群之间存在显著差异。致病变异率在不同祖先人群间存在的显著差异,可能由在变异数据库中确定致病变异时存在的偏差所导致,或由祖先群体之间潜在的疾病患病率差异所导致。
为全面地解析参与者的致病性和可能致病性变异,研究团队将罕见、预测功能丧失(pLoF) 变异纳入已知致病变异的先前分析中,并重点研究了38个特定基因。结果显示,共发现1,114 个变异,其中562个移码变异、112个剪接受体变异、100个剪接供体变异和340个终止增益变异;许多pLoF变异与VIP数据库中的致病/可能致病变异重叠。此外,不同血统组之间pLoF变异的差异小于致病/可能致病变异的差异。
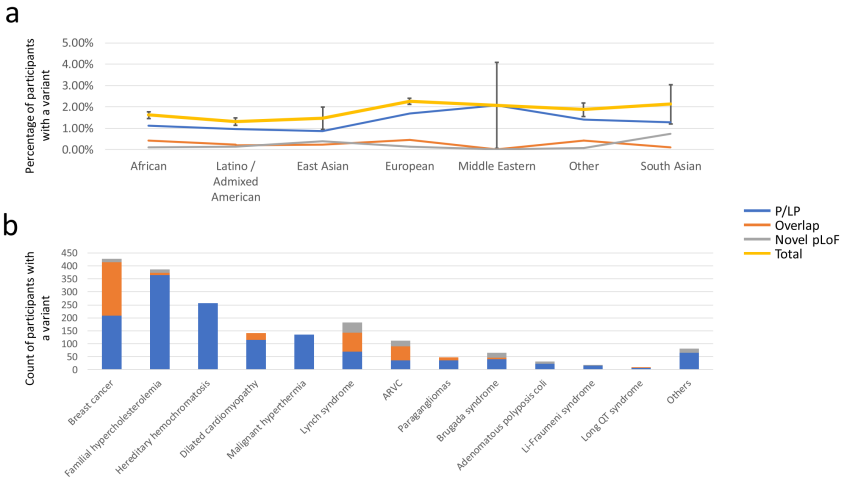
图1. 祖先驱动的致病变异
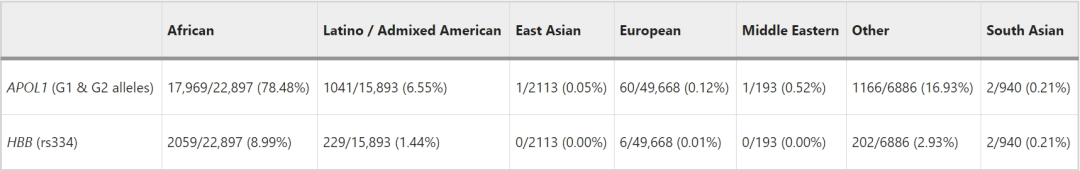
接下来,研究团队评估了具有祖先差异的已知变异能否在All of Us队列中重现,检测了与镰状细胞病相关HBB-rs334突变的频率,以及APOL1 G1和G2等位基因。结果证实了预期的祖先差异,非参考等位基因在22,897名非洲血统的参与者中出现17,969次,在49,668名欧洲血统的参与者中出现68次。
表1. 已知祖先差异基因中的非参考样本计数
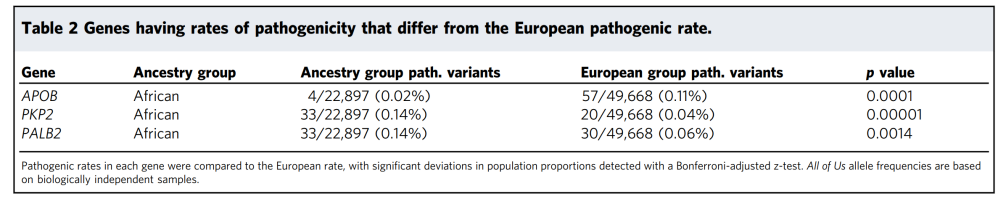
为了解在不同祖先群体之间具有不同致病变异率的基因,研究团队将所有基因的致病率与欧洲祖先群体的致病率进行归一化处理,发现有几个基因显示出显著差异。除已知的HFE和PALB2基因的差异外,该研究首次报道了PKP2基因的差异。此外,APOB基因显示出的差异可能表明某些祖先群体中遗传疾病发病率来源发生改变。上述基因水平的差异是未来健康差异研究的重点。
表2. 与欧洲亚组致病变异率不同的基因
研究团队将All of Us队列的致病发现率与gnomAD等大型队列进行了比较,图2显示了按基因细分的All of Us和gnomAD队列阳性率之间的相对差异。总体而言,上述数据源之间存在高水平一致性;但除欧洲祖先群体外,其他祖先群体的基因水平致病率存在一些明显差异。
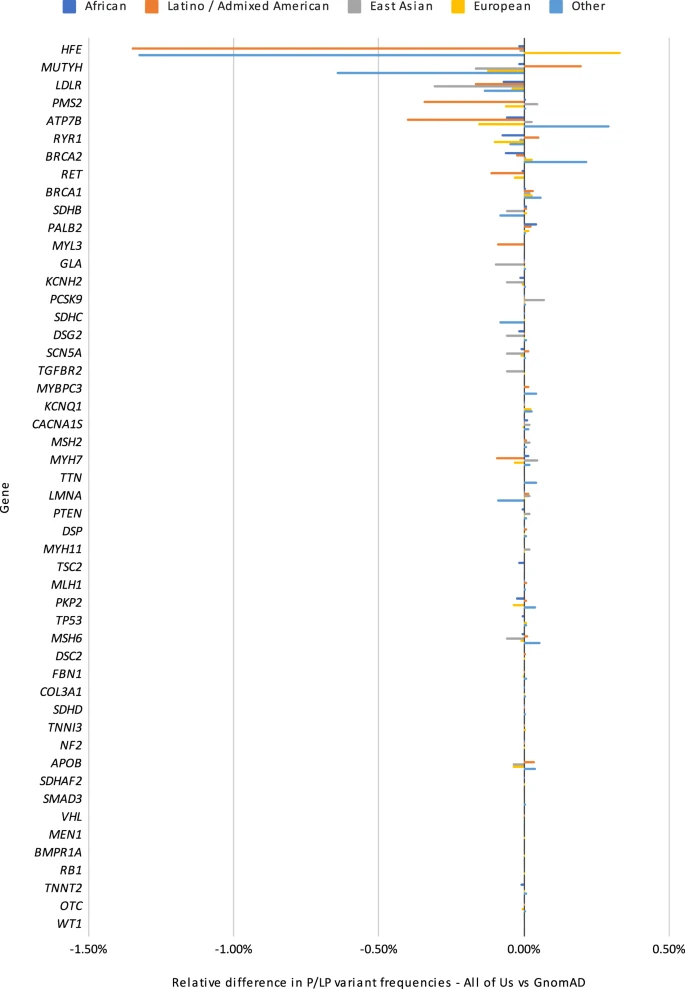
图2. All of Us与gnomAD的相对阳性率。
评估潜在的参与者选择效应
为进一步了解已知遗传疾病参与者中是否自愿选择参与All of Us计划,这可能影响孟德尔遗传病的致病变异计数,研究团队选择了与疾病相关的4个罕见变异和4个常见变异,并检查了其相对于GnomAD数据库的等位基因频率。为最好地匹配All of Us祖先群体,GnomAD欧洲群体被视为芬兰人、非芬兰人和德系犹太人的组合。对于上述变异,研究团队观察到GnomAD和All of Us等位基因频率之间密切匹配;只有常见的HFE rs1800562等位基因存在显著差异,差异为6%。
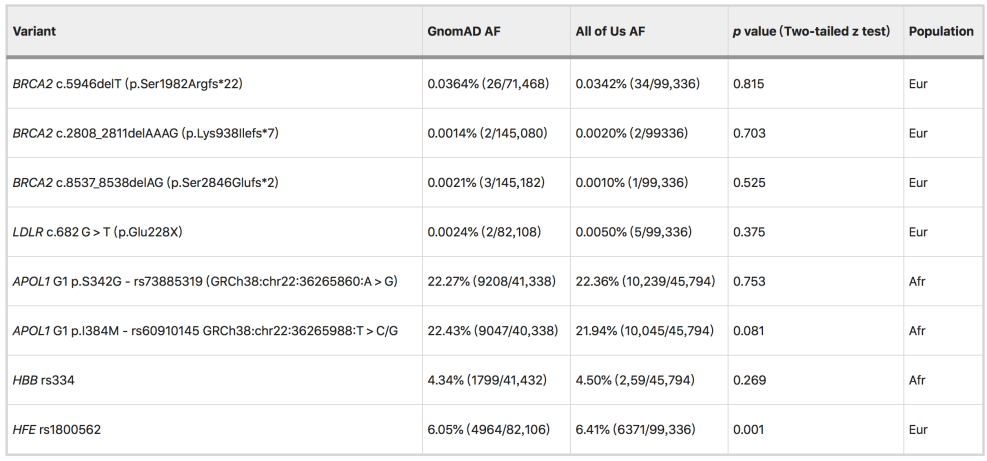
图3. GnomAD特异变异等位基因频率比较
特定遗传因素的富集分析
当按疾病领域细分时,与健康相关的主要发现是乳腺癌、家族性高胆固醇血症、扩张型心肌病和遗传性血色素沉着症。为进一步了解这些发现与参与者现有健康信息的关系,研究团队利用All of Us云计算平台提供的额外数据资源进行深入分析,其包括参与者的健康信息,以及参与者回答的调查问题。
以乳腺癌为例,研究团队分析了云计算平台上的表型信息如何与基因发现相匹配。如果参与者有“女性乳房恶性肿瘤”或“乳房恶性肿瘤”病症代码,或参与者在“医生或保健提供者是否告诉过您患有或曾经患有以下癌症”的问题中回答“乳腺癌”,则选择该样本。结果显示,乳腺癌队列共有8,603名参与者,其中1,653人拥有WGS数据。上述结果表明,乳腺癌患者P/LP变异存在富集,证明了使用All of Us参与者水平数据来富集特定遗传因素的能力。
结 语
综上所述,All of Us收集的多样化队列将是推进精准医学的丰富资源。在该研究中,研究团队检查了云计算平台数据的致病变异率,发现具有不同祖先的参与者群体之间存在显着差异,这种变异可能是多种因素的结果。未来的工作将逐步揭示All of Us多样化队列的变异注释是否会对这种致病变异的确定偏差产生影响,未来针对非欧洲参与者进行临床解释的研究分析十分必要。
论文原文:
Venner, E., Patterson, K., Kalra, D. et al. The frequency of pathogenic variation in the All of Us cohort reveals ancestry-driven disparities. Commun Biol 7, 174 (2024). https://doi.org/10.1038/s42003-023-05708-y
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




#祖先驱动# #致病变异率#
12