
记1例MDS快速转AEL的诊疗经过
2023-05-24 徐益恒等 昆明医科大学第二附属医院 “检验医学”公众号 发表于上海

通过该病例,大家都深刻认识到,检验不但能为疾病诊疗提供客观证据,还能带领临床有的放矢,认识疾病,治疗疾病。
前 言
本案例中患者以乏力、发热起病,血常规发现贫血及血小板减少就诊,入院后各种检验检查多管齐下,感染检测一无所获,临床医生一筹莫展,骨髓穿刺结果变得尤为期待。不做不知道,一做吓一跳,骨髓涂片上的一群体积大,胞浆少,染色质细的分类不明细胞达11%(当时考虑淋巴瘤细胞),并可见吞噬血细胞现象,大量幼红细胞形态异常,由此开启了一段检验与临床的你来我往的沟通、合作的呕心沥血过程,现代血液形态学、免疫学、遗传学、分子生物学检查大显身手的陆续登场,检验检查手段的运用与疾病的快速进展从来没有停歇,但是患者的病情起起落落,特别揪心。
虽然最终诊断并非最初怀疑的淋巴瘤,但是我们并没有因此而沮丧和挫败,反而更觉得形态学的巨大作用,血液病几乎都是始于形态学也终于形态学。该病例最终诊断为TP53双突变型MDS伴复杂遗传学改变,并发噬血细胞综合征,自身免疫性溶血性贫血,快速进展为罕见的急性红系白血病(AEL)。通过该病例,大家都深刻认识到,检验不但能为疾病诊疗提供客观证据,还能带领临床有的放矢,认识疾病,治疗疾病。
案例经过
患者男性,47岁,乏力1月余、发热2周、发现血细胞异常1周于2020年8月10日就诊。2020年7月无明显诱因出现乏力、四肢酸软,未重视及诊治。8月初出现发热,最高体温38.6℃,自行物理降温及服用抗病毒颗粒、克感敏治疗无明显好转,且伴四肢皮肤自发散在瘀斑。
至当地社区行血常规提示WBC 6.02×109/L,HGB 72g/L,正细胞正色素,PLT 39.02×109/L;铁三项:铁蛋白1232.6ng/mL,铁8.3umol/L,转铁蛋白1.98g/L,外院补铁(具体不详)治疗无好转。
近半年体重下降5Kg,近1月下降约2kg。以“1.贫血、血小板减少查因;2.发热查因”收住院,查体:T: 39.6℃,P: 90次/分,R: 20次/分,BP: 120/80mmHg。贫血貌,巩膜无黄染,左膝关节、右上臂、右手背可见片状瘀斑,无皮疹,胸部叩诊清音,双肺呼吸音清晰,全身浅表淋巴结无肿大,胸骨无压痛。肝脾肋下未触及。
入院后相关检查:2020-8-12骨髓细胞学示(右髂后):增生明显活跃(Ⅱ),髓象粒、红、巨系增生,不典型(幼稚样)淋巴占11.0%,可见吞噬血细胞现象,外周血涂片中可见不典型(幼稚样)淋巴细胞占1%。
2020-8-13左髂前骨髓细胞学:骨髓增生减低(Ⅳ),髓象粒、红、巨系增生减低,幼淋样细胞占4.0%,外周血涂片中亦可见不典型(幼稚样)淋巴细胞占2%。2020-8-18胸骨穿刺骨髓细胞学:增生重度减低,分类计数类似2020-8-13。以上三次骨髓细胞学原始粒细胞比例均为0,但是对幼红细胞的形态异常都进行了描述,而没有分类计数异常比例。
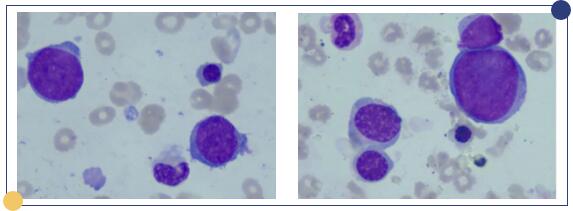
图1:(左)2020年8月12日骨髓涂片;(右)2020年8月14日骨髓涂片
均可见一类体积较大,胞浆较少的不典型(幼淋样)细胞,疑为淋巴瘤细胞
骨髓活检示:骨髓部分区域伴出血,无法准确评估增生程度,粒、红、巨三系可见伴幼稚细胞略增多(据形态学、外周血全血细胞减少改变,需排外营养性疾病、病毒感染、慢性炎症、药物、重金属中毒、铜缺乏、过度饮酒,再考虑骨髓增生异常综合征),骨髓活检免疫组化:免疫组化:CD34个别+,CD117个别+,MPO粒系细胞+,Lysozyme散在分布+,CD42b巨核细胞+,CD20个别+,CD3少量+,CD56-,GPA红系细胞+,CD138浆细胞+。
图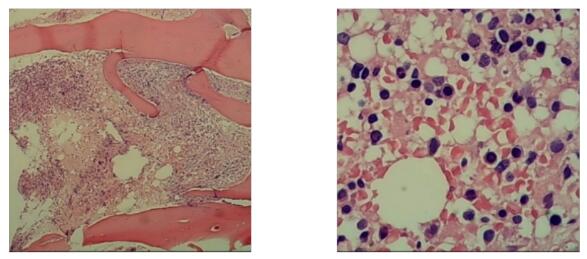
图2 2020年8月12日骨髓活检
流式免疫分型(淋巴细胞系列抗体)未见异常;
髓系流式免疫分型 髓系原始细胞比例不高(1.6%),表型未见明显异常;粒系比例减低,以晚幼粒细胞及之后阶段为主;红系比例增高,部分细胞CD36表达减弱或缺失;单核细胞和淋巴细胞未见异常表型;请结合临床及其它检查。 各系比例如下:

图3 2020年8月12日流式细胞术检查结果
血常规三系进行性下降,以血红蛋白下降明显,最低为45g/L;间接胆红素25.1umol/L↑,LDH 2529U/L、铁蛋白>1500ng/mL; NK细胞活性7.52%;可溶性CD25为21431pg/mL;Coomb’s试验阴性;自身免疫全套阴性。
染色体核型分析示:可见克隆性异常-Y, der(2), -12, add(20q) [10];
MDS二代测序示:TP53基因双突变;突变位置均位于Exon8,氨基酸序列分别为p.R273H(突变频率36.1%)和p.V272M(突变频率37.7%)。
检验案例分析
(1)第一阶段住院:患者中年男性,以乏力、继而高热起病,病史短。临床以血细胞减少及不明原因发热,进行骨髓穿刺,首先做骨髓涂片细胞学检查,当时接到主管医生的求助,顿感情况紧急,立即进行染色显微镜下观察,发现一类体积较大,胞浆较少,染色质细致均匀的细胞,比例竟达11%,并且找到嗜血细胞。
仅凭形态学,一下子确实难于准确归类。考虑到病史及临床表现,我们马上反馈,该患者要考虑淋巴瘤累及骨髓的可能性,接下来必须做骨髓活检+免疫组化、流式免疫分型。结果证实这些体积较大的细胞不是淋巴细胞,也不是髓系原始细胞(粒、单、巨核),并没有更进一步明确的归类,此时我们形态学人员也没有想到会是原始红细胞。
回到骨髓细胞形态学,该患者以红系发育异常为主,粒系、巨核系也可见发育异常,免疫表型检查排除了异常淋巴细胞表型,诊断陷入迷茫。骨髓细胞形态学发育异常主要见于MDS,但是患者多次骨髓穿刺,并没有髓系原始细胞增多,这样绕不开病史最低要求4个月的这一主要诊断标准,还有办法吗?回答是肯定的,有!
血液肿瘤的诊断从20世纪90年代就进入MICM时代,到此,我们已经有形态学、免疫学的检查结果,接下来遗传学、分子生物学检查或许找到一线希望,病人情况紧急,伴有噬血及溶血,发热及血小板减少时刻在危及患者生命。染色体核型示:可见克隆性异常-Y,der(2),-12,add(20q)[10]。MDS二代测序示:TP53基因双突变;突变位置位于Exon8,氨基酸序列分别为p.R273H(突变频率36.1%)和p.V272M(突变频率37.7%)。这些客观检查结果,对临床的诊断非常有用,预示高危的MDS。
(2)第二阶段住院:本次入院后骨髓细胞学涂片,有核细胞总数、红系比例、原始红细胞比例均较前增多,该患者原红的形态,较正常的原红,有很大不同,体积大小不均,类圆形为主,胞浆少,浆色较浅,染色质较细,核仁数量少,部分还有假性颗粒。在流式免疫分型的帮助下,才可以和髓系原始(粒、单、巨核)及淋巴加以区分,红系最高时达76%,原红最高时达29.5%,并且原始红细胞PAS染色多数为颗粒状阳性,支持急性纯红系白血病(AEL)诊断。
(3)MDS是较复杂的髓系肿瘤之一,实验室检查对血液系统疾病,尤其是血液肿瘤具有重要地位。我们实验室对形态学非常重视,这为血液系统疾病的诊断奠定了基础。形态学是MDS诊断的基础之一,但形态学分析的工作量也大,难度也很大,且形态学诊断的主观性也不适应精准医学的要求,单纯关注形态学无法满足MDS临床诊断和分型的需要,需要建立区域性的、完备的、有质量的现代骨髓检查技术中心,而不是各自为阵,才能彻底满足类似本例这样疑难急重患者诊疗的需要。
临床角度分析
首先患者发热、全血细胞减少、铁蛋白升高、NK细胞活性下降、sCD25升高、骨髓细胞学见到噬血现象,LDH升高、胆红素升高;结合以上检查需警惕噬血细胞综合征(HLH);其次患者髓系免疫分型示未见原始细胞,但红系比例增高,部分细胞CD36表达减弱或缺失;结合患者病史及相关检查检验,排除淋巴瘤骨髓累及。
患者骨髓活检提示MDS,但结合病史血细胞减少小于4月,基因及染色体核型未回,按2019年《MDS中国诊断与治疗指南》最低诊断标准,MDS依据不足。初步诊断考虑:1.不明原因发热继发血细胞减少伴噬血现象;2. MDS待观察;予抗感染、地塞米松等治疗后血小板波动在(22~26)×109/L,白细胞及血红蛋白上升、体温正常。
出院后继续服用“甲泼尼龙20mg qd”,至2020年9月初减量至16mg qd,2020年10月初减量至12mg qd。复查血常规提示白细胞正常,血红蛋白105-115g/L,血小板波动在(21~30)×109/L。继续检查,同时等待检查结果。
随后染色体核型分析示-Y, der(2), -12, add(20q) [10];MDS二代测序示:TP53基因双突变;患者激素治疗有效,是否合并结缔组织病、HLH。但结合遗传学及分子生物学检查,已经符合MDS标准,但是激素有效,如何解释,积极治疗?、BMT?、化疗?、等待?做免疫相关检查,但是都是阴性?又怎么办?,在我们这里患者予继续等待。
2020-10-12(2个月以后)因再次血小板减少伴发热第二次入院。复查血常规白细胞从6.5×109/L降至1.75×109/L,血红蛋白波动于68-80g/L,血小板波动于(13~20)×109/L,髂后骨髓细胞学提示:骨髓增生活跃(Ⅲ),G=38.0%,E=52.0%,G/E=0.73:1。粒系增生活跃,比例减低,可见少分叶、异常分叶、双核、环形核及类巨变,部分颗粒减少。红系增生活跃,比例增高,以中、晚幼红细胞为主,原始红细胞增多,部分幼红细胞呈类巨变,核畸形,多核,胞质空泡变等异常,成熟红细胞大小不均。

图4 2020-10-12骨髓细胞涂片
红系比例增高,原始红增多,幼红细胞,粒细胞均可见发育异常
骨髓活检示:本次骨髓活检增生极度活跃(90%),可见一类胞体大的细胞,骨髓幼稚红系细胞增多,需要鉴别骨髓增生异常综合征及纯红系白血病;骨髓组织免疫组化:骨髓活检中可见一类胞体大的细胞为幼稚红系细胞,表型为:CD34- , CD117 + , MPO- , GPA少量+ , CD71+ , TDT-, CD19-, CD3-, CD42b-, CD61-。
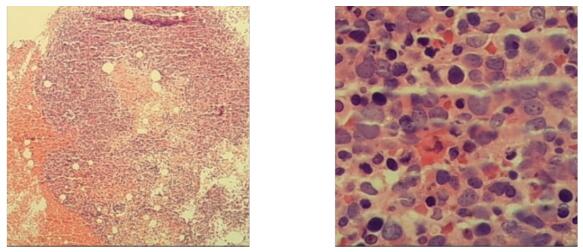
图5 2020年10月13日骨髓活检
髓系流式免疫分型:髓系原始细胞比例不高,表型未见明显异常;粒系比例减低,以晚幼粒细胞及之后阶段为主;红系比例明显增高,占有核细胞的59.59%,部分细胞FSC及SSC增大,部分细胞CD36表达减弱或缺失,其中CD117+CD105+的早期红细胞占有核细胞的15.17%;单核细胞和淋巴细胞未见异常表型;各系比例如下:
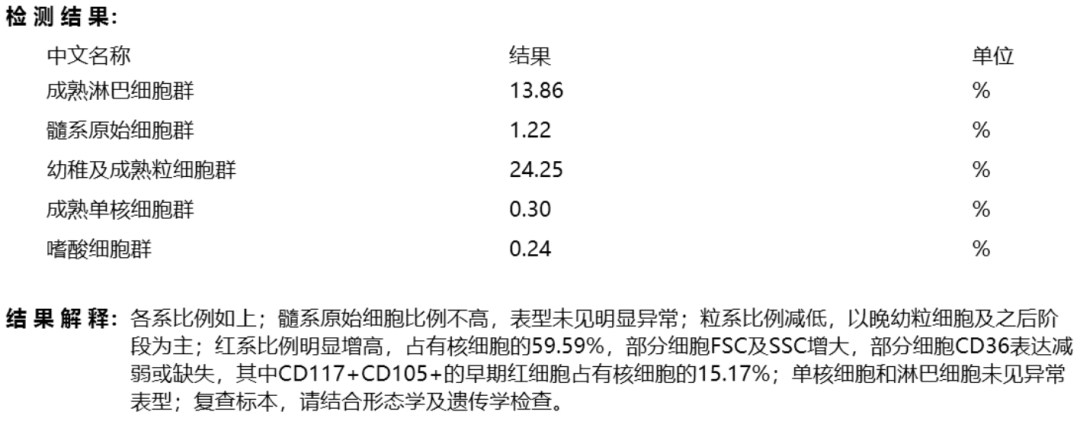
图6 2020年10月13日骨髓流式免疫分型
Coomb’s试验阳性;LDH正常,铁蛋白>1500ng/mL。
病情变化:患者前期MDS诊断明确,激素治疗有效。但病情短期内出现反复及加重。第二次的检查结果出现更加明显的病态造血,原始红细胞比例增多。结合病史,修正诊断为急性红白血病(伴MDS转化)。
2020年10月20日予阿扎胞苷+CAG方案化疗:阿扎胞苷100mg, d1-d7;阿糖胞苷30mg, d1-d14;阿柔比星20mg, d1-4;G-CSF 400ug d0至白细胞>20×109/L。化疗期间及化疗后骨髓抑制,合并皮肤黏膜出血、血尿、便血。停化疗后1月后复查骨髓细胞学,原幼红细胞比例20.5%,评估未缓解。2021年1月20日患者因突发颅内出血去世。
患者系中年男性,以乏力、发热起病,贫血、血小板减少,迅速进展为全血细胞减少,骨髓形态学可见胞体大的分类不明细胞,吞噬血细胞现象,到骨髓有核细胞数量、红系比例及早期红细胞比例均动态增加;红系形态异常变化明显;细胞遗传学及分子生物学可见复杂染色体核型、双突变型TP53;最终转化为急性红白血病。且在病程中反复发热、合并溶血及严重的出血,病情进展快。
在该病例中,患者初诊时在缺乏克隆性造血证据的情况下,激素治疗有效,病程相对较短,未达到MDS最低诊断标准。后期患者很快失去激素疗效,结合后续报告提示双突变TP53和复杂染色体核型等,MDS诊断明确。回顾该病例,若治疗时机提前,治疗策略更积极,可能改善患者最终临床结局。
知识拓展
按2022年WHO第5版分类MDS分类,分为具有明确的遗传学异常和明确的形态异常两大类亚型。双等位基因TP53失活的MDS (MDS-biTP53)是具有明确遗传学异常之一,这一类患者骨髓和外周血原始细胞<20%,遗传学异常通常复杂,存在两个或多个TP53突变。
当检测到两个或多个TP53突变时,通常会影响两个等位基因,并且可以被认为是多重打击状态。超过90%的MDS-biTP53患者具有复杂的、大多数非常复杂(>3)的核型,因此在IPSS-R中被认为具有非常高的风险。需要进一步的研究来确定biTP53本身是否是AML特有的,这是未来版本修订中是需要考虑的。尽管如此,已发表的数据表明,出于治疗考虑,MDS-biTP53可被视为AML相似的疾病。
在不同的研究中,AEL患者最常见的突变是TP53(31%), NPM1 (14%)和DNMT3A (11%)。不同人种、年龄的患者,存在突变频率的差异。TP53突变患者预后较差,而NPM1突变患者5年生存率可达88%。
成人AEL的基因突变频率处于MDS和AML之间,FLT3和NPM1突变低于非红系AML但高于MDS,但MDS相关的突变,如SF3B1和ASXL1在AEL少见,AEL常见表观遗传学调控基因ATM、CREBBP、ATRX1、SETD2的突变,Ras通路基因以NF1为常见,转录调控基因以IKZF1和WT1突变常见,成人AEL罕见PPM1D和SRSF2基因突变。
TP53突变是AEL的特征,高达37.9%。研究发现,红系发育调控基因特别是GATA1、KLF1、NFE1表达异常,它的异常是由表观修饰基因DNMT3a和TET2突变相互作用驱动的,这些修饰基因突变致甲基化修饰紊乱进而影响谱系特异性转录因子表达。
本例患者系TP53双突变及复杂染色体核型,临床进展较快,OS仅为5月。因此需强调双等位基因TP53突变在这种侵袭性AML类型中的核心作用,不能因为激素疗效影响本病的治疗。药物敏感性与基因突变类别有关,去甲基化剂地西他滨对TP53、Bcor突变型AEL有效,该例患者治疗时将阿扎胞苷调整为地西他滨可能是更优选择。
案例总结
该例患者就诊时骨髓细胞形态学可见胞体大细胞,骨髓活检考虑MDS可能,流式免疫分型提示红系异常,淋巴瘤流式免疫分型未见异常,且合并TP53双突变和复杂染色体核型,局限在MDS诊断标准,整合资料时不准确,最终患者转为急性红白血病,OS为5月。
因该例患者红系细胞形态学特征不典型,且AEL临床少见,那么此时强大的检验支持即很重要;因此,加强临床与检验的沟通,将MICM分型相关检查资料综合分析,使诊断更精准,得到及时治疗,提高患者预后。
在MDS诊断中,形态学是基础,但是2022版本的指南,将基因异常的重要性大大提升。目前在诊断时我们都完善了MDS二代测序,当拿到一份NGS检测报告,我们应该怎么看待检测结果呢?MDS二代测序检测中,基因的突变不能用于诊断,多用于预后判断。尽管约90%的MDS患者存在有细胞遗传学和(或)基因异常,也就是说可以找到克隆性证据,结合2022年WHO中MDS现有分类,发现SF3B1基因突变与骨髓环状铁粒幼红细胞增多有较特异的相关性和双等位基因TP53失活的MDS,可以用于分型诊断。
参考文献
[1] Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Barreto de Oliveira Araujo I, Berti E, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/ Dendritic Neoplasms. Leukemia. 2022 Jul;36(7):1703-1719.
[2]Cristina Di Genua , Claus Nerlov.To bi or not to bi: Acute erythroid leukemias and hematopoietic lineage choice. Exp Hematol . 2021 Feb 16;S0301-472X(21)00083-7. doi: 10.1016/j.exphem.2021.02.006.
[3]Ilaria Iacobucci,et al. Genomic subtyping and therapeutic targeting of acute erythroleukemia. Nat Genet . 2019 Apr;51(4):694-704. doi: 10.1038/s41588-019-0375-1.
[4]Ilaria Iacobucci, Chunxu Qu, et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood. 2021;137(12):1628-1640. doi: 10.1182/blood.2020009103.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






也许将来以tp53突变来命名一种血液病更合理,可以更早干预
32
认真学习了
40