
NEJM述评:达比加群引起的出血风险不高于华法林
2013-03-21 shumufeng 丁香园

达比加群VS华法林引发的颅内出血和胃肠道出血事件 美国食品和药品监督管理局(FDA)于2010年10月批准口服抗凝药达比加群([abigatran] Pradaxa,Boehringer Ingelheim)上市。此后的几年中,FDA不良事件报告系统(FAERS)收到许多使用该药导致严重和致死性出血事件的报告。达比加群是一种抗凝药,因此导致出血事件在意料之中,但报告发生率
达比加群VS华法林引发的颅内出血和胃肠道出血事件
美国食品和药品监督管理局(FDA)于2010年10月批准口服抗凝药达比加群([abigatran] Pradaxa,Boehringer Ingelheim)上市。此后的几年中,FDA不良事件报告系统(FAERS)收到许多使用该药导致严重和致死性出血事件的报告。达比加群是一种抗凝药,因此导致出血事件在意料之中,但报告发生率远大于华法林[warfarin]相关并发出血报告发生率,华法林是在达比加群被批准前近60年里一直在使用的抗凝药。与以下表述不同的是,支持达比加群批准的对照试验(长期抗凝治疗的随机化评估研究[RE-LY]),在非瓣膜房颤患者中比较华法林与达比加群[1]的安全性,结果显示两药出血风险相似。
达比加群上市后的出血报告导致医学刊物以及主流媒体对监督管理局批准该药产生了广泛质疑。它们引证FDA不良事件报告系统(FAERS)出血事件报告作为质疑达比加群说明书中描述的获益-风险图的理由。但可能影响到报告率的重要因子却不被重视,例如达比加群新颖性(相对于已为大家接受的华法林)和媒体中新药报道的力度等,均可能大大影响不良事件的报道时间和报道方式。
RE-LY试验纳入有非瓣膜房颤或卒中风险的患者(至少存在一种风险因子)。结果显示,150 mg剂量每天2次达比加群治疗在减少卒中和全身性栓塞合并发生率率方面优于华法林(每100患者-年1.1 VS 1.7)。与华法林相比,达比加群治疗组患者的血栓栓塞和出血性卒中发生率均更低,但华法林治疗组死亡率较低(3.6 VS 4.1每100患者-年)。150 mg剂量达比加群治疗组和华法林治疗组患者的主要风险水平,出血风险等相似(重大出血事件发生率分别为3.3和3.6每100患者-年)。(RE-LY研究中重大出血事件定义为血红蛋白浓度降低至少2 g/dL,需要输血或红细胞压积至少2单位,或在关键性部位或器官出现症状性出血。)
虽然与华法林组相比,达比加群组重大胃肠道出血事件发生频率更多(1.6 VS 1.1每100患者-年),颅内出血事件发生率却较低(0.3 VS 0.8每100患者-年)。达比加群(150-mg剂量)治疗减少卒中和全身性栓塞率优于华法林,而临床上显著出血发生率二者相似,这些结果促使FDA批准达比加群。
RE-LY试验已清晰地显示达比加群有严重副作用——出血,因此药品批准后将建立出血事件报告制度,但报告数量如此之高以至于FDA不得不启动不良事件报告系统 (FAERS)接收处理自发性报告的评述。我们注意到上市后达比加群的使用可能不同于在RE-LY试验中的使用(如,不同的患者群,给药,同时用药,和肾受损程度)或没有根据患者肾功能状况进行正确的给药剂量调整。
如同常见的自发性报告,出血事件报告一般不述及患者的风险因子、年龄、肾功能、或死亡原因等信息。对一小部分肾功能受损患者,达比加群使用剂量未予减少。总之,病例综述未鉴定出任何尚未认识到的出血风险因子,也没有发现任何与其标签说明一致的达比加群禁忌证指示因子。
因此,我们考虑了以下可能性:已经接受达比加群治疗的患者人群中出现的意料之外的出血事件报道高发生率预示着正在接受达比加群治疗的患者可能会出现比华法林治疗更多的出血事件。这一趋势的推动力来自美国以外监管当局和已发表病例报告和安全性通讯,以及达比加群新上市等待因素。
我们知道有关一款药的物不良事件或合法活动的新闻可能增加其报告率。我们也知道新上市药品,单凭其新颖性即可引发较高的不良事件报告率;报告率随时间减少(即所谓的Weber效应)。因此,华法林,这款上市已近60年并广人知的药物引起出血等不良事件的报告率比风险相似的新药低得多。虽然FDA也承认有关达比加群的非预计高数量出血报告很可能也存在上述因素的原因,但我们依然按照FDA实践标准在2011年12月,面向保健医生和患者发出一份药物-安全性通讯以传达关于达比加群可引发出血的信息。
显然,FDA药物监察的神经使命促使其了解各种各样的相关因子,其中许多因子可能与药理学本身无关,然而可能会影响药品上市后的不良事件报告。达比加群案例中,我们试图确定有关出血报告如此之多是否反映在上市后情境中相对于华法林出血风险是否真实增加。我们利用保险-索赔数据[insurance-claim data]和来自FDA Mini-Sentinel数据库的数据,来比较达比加群和华法林引起的出血率。
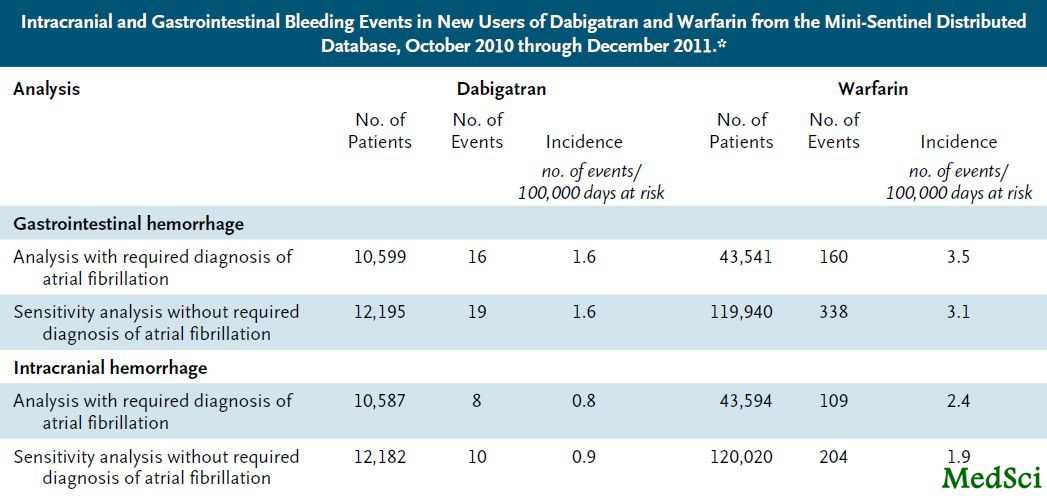
这个数据库可用来评估出血诊断发生率和入选人群使用的药物。我们查询了数据库从 2010年10月19日(达比加群批准日期)至2011年12月31日这个阶段的记录,对使用达比加群或华法林的颅内和胃肠道出血的住院病人的诊断编码加以鉴别(见表)。我们发现在目标时间段内,伴随达比加群使用患者出血率表现不高于华法林。
Mini- Sentinel分析有其局限性,包括缺乏对混杂变量的校正和缺乏详细医疗记录评述(以验证诊断代码是否反映真实出血发生),为解决这些局限性,目前我们正在进行两项基于方案的评估,利用来自Mini-Sentinel的索赔数据和其他索赔数据库,校正混杂因子。
我们认为有关达比加群的出血报告数量如此之大,这是一个显著的刺激报告案例。在本例中,这类报告对临床实践中使用达比加群VS华法林引起的出血事件发生率进行了扭曲的评估。Mini-Sentinel 评估提示达比加群有关出血率不高于华法林,这一发现与RE-LY结果一致。虽然有些人已注意到对达比加群的抗凝效应缺乏可供利用的逆转剂,这成为其使用的重要局限性,然而来自RE-LY研究的数据给出了关于出血事件安全性的保证。我们相信当按照说明加以应用时,达比加群可产生重要健康获益。
作为常规的FDA不良事件报告系统(FAERS)上市后监察渠道,针对Mini-Sentinel和其他索赔数据库的进一步分析正在进行中。
针对达比加群的最新建议包括:
● 鉴于肾功能损害是达比加群酯发生出血风险的危险因素:开始治疗前,所有患者都要评估肾功能,以排除重度肾功能损害;治疗期间,如果怀疑有肾功能减退的临床情况,要常规复查肾功能;老年患者(>75岁)或中度肾功能损害的患者,至少每年复查1次肾功能。
● 达比加群酯不宜用于血流动力学有明显风湿性心脏瓣膜病表现(特别是二尖瓣狭窄)的患者和有人工心脏瓣膜的患者。
达比加群出血风险和肾功能评估:
依据达比加群酯上市后严重出血事件的报告,以及老年患者和出血高危患者或肾功能损害患者的使用情况,对产品专论的内容做了更新。目前更新后的内容包括,建议对考虑使用或已经使用达比加群酯治疗的患者要进行肾功能评估,具体内容如下:
● 开始用达比加群酯治疗前,所有患者都要通过计算肌酐清除率(CrCl)评估肾功能,以排除重度肾功能损害(即CrCl<30 ml/min)。
● 达比加群酯治疗期间,如果临床上有怀疑肾功能快速减退或迅速恶化的情况(如血容量不足、脱水以及同时使用某些药物),要复查肾功能。这些临床情况可能导致达比加群酯的暴露量增加。
● 老年(>75岁)患者或中度肾功能损害(CrCl 30-50ml/min)的患者,要通过计算肌酐清除率评估肾功能,每年至少1次。
给医疗专业人员的建议:
● 达比加群酯禁用于重度肾功能损害(CrCl<30 ml/min)的患者。
● 出血高危患者也不要使用达比加群酯。
● 要对患者进行临床监测,注意有无出血或贫血的表现。
● 如果发生严重出血,应停用达比加群酯,并查明出血部位。
心脏瓣膜疾病的患者:
达比加群酯的安全性和有效性尚未在血流动力学上有明显风湿性心脏瓣膜病表现(特别是二尖瓣狭窄)的患者或有人工心脏瓣膜的患者中进行研究。没有数据支持达比加群酯在安装人工心脏瓣膜的患者(无论是否有心房纤颤)中具有充分的抗凝效果,因此,达比加群酯不宜用于血流动力学上有明显风湿性心脏瓣膜病表现的患者或有人工心脏瓣膜的患者。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



