ATVB 复旦大学魏园园团队揭示miR-223-3p通过抑制Ripk3介导的巨噬细胞程序性坏死减缓动脉粥样硬化晚期发展的作用机制
2023-11-28 论道心血管 论道心血管 发表于上海

研究揭示了miR-223-3p通过抑制Ripk3介导的巨噬细胞程序性坏死减缓动脉粥样硬化易损斑块形成的作用机制。
动脉粥样硬化的病理机制极其复杂且发病过程不可逆,有效地治疗及预防动脉粥样硬化还面临着巨大挑战。易损斑块的形成是晚期动脉粥样硬化的典型标志,而坏死核心的存在是斑块易损的主要原因之一。因此,进一步了解坏死核心形成的分子机制并寻找新的靶向抑制坏死核心的策略对于稳定易损斑块具有非常重要的意义。
在动脉粥样硬化的发展过程中,尤其是晚期易损斑块中存在多种形式的细胞程序性死亡。除了细胞凋亡,其他形式的细胞死亡也不同程度地影响了斑块的形成和稳定性。但是,每种细胞死亡形式对动脉粥样硬化的发展和斑块易损性的贡献是否一致,以及是否存在某种主要的细胞死亡形式加速了斑块破裂还不得而知。因此,探究清楚斑块中存在的主要细胞死亡形式对通过靶向细胞死亡稳定易损斑块的治疗策略极其重要。
2023年11月16日,复旦大学基础医学院魏园园课题组和德国慕尼黑大学医院心血管病预防研究所Andreas Schober课题组合作在Arteriosclerosis, Thrombosis, and Vascular Biology杂志在线发表题为“miR-223-3p Prevents Necroptotic Macrophage Death by Targeting Ripk3 in a Negative Feedback Loop and Consequently Ameliorates Advanced Atherosclerosis”的研究论文,揭示了miR-223-3p通过抑制Ripk3介导的巨噬细胞程序性坏死减缓动脉粥样硬化易损斑块形成的作用机制。

首先,研究人员探究了人及小鼠动脉粥样硬化斑块中存在的主要细胞死亡形式。结果显示,相比于早期稳定斑块,在人的不稳定斑块和经24周高胆固醇喂食的小鼠主动脉斑块中,程序性坏死的效应蛋白磷酸化MLKL显著表达上调,而研究人员并未发现细胞焦亡和凋亡效应蛋白的明显变化,提示RIPK3介导的程序性坏死在不稳定斑块中被显著激活。
为了进一步寻找抑制巨噬细胞程序性坏死的分子机制,研究人员结合Targetscan预测了RIPK3上游的负向调控分子miR-223-3p,并通过荧光素酶实验证明了RIPK3 3′ UTR中存在miR-223-3p的靶位点。在程序性坏死的诱导条件下,Mir223基因缺失的小鼠骨髓巨噬细胞和转染了miR-223-3p抑制剂的人巨噬细胞U937的死亡率均显著上升。相应的,Mir223的敲除上调了Ripk3的蛋白表达及其磷酸化水平并增强了MLKL的磷酸化,进而沉默或缺失Ripk3阻止了由Mir223缺失导致的细胞程序性坏死率的上升(图1)。研究人员进一步通过荧光素酶报告基因实验发现,Ripk3可通过上调转录因子C/EBPβ进而增强miR-223的转录。因此,Ripk3/C/EBPβ/miR-223-3p组成了调控泡沫巨噬细胞程序性坏死的负反馈调控网络。
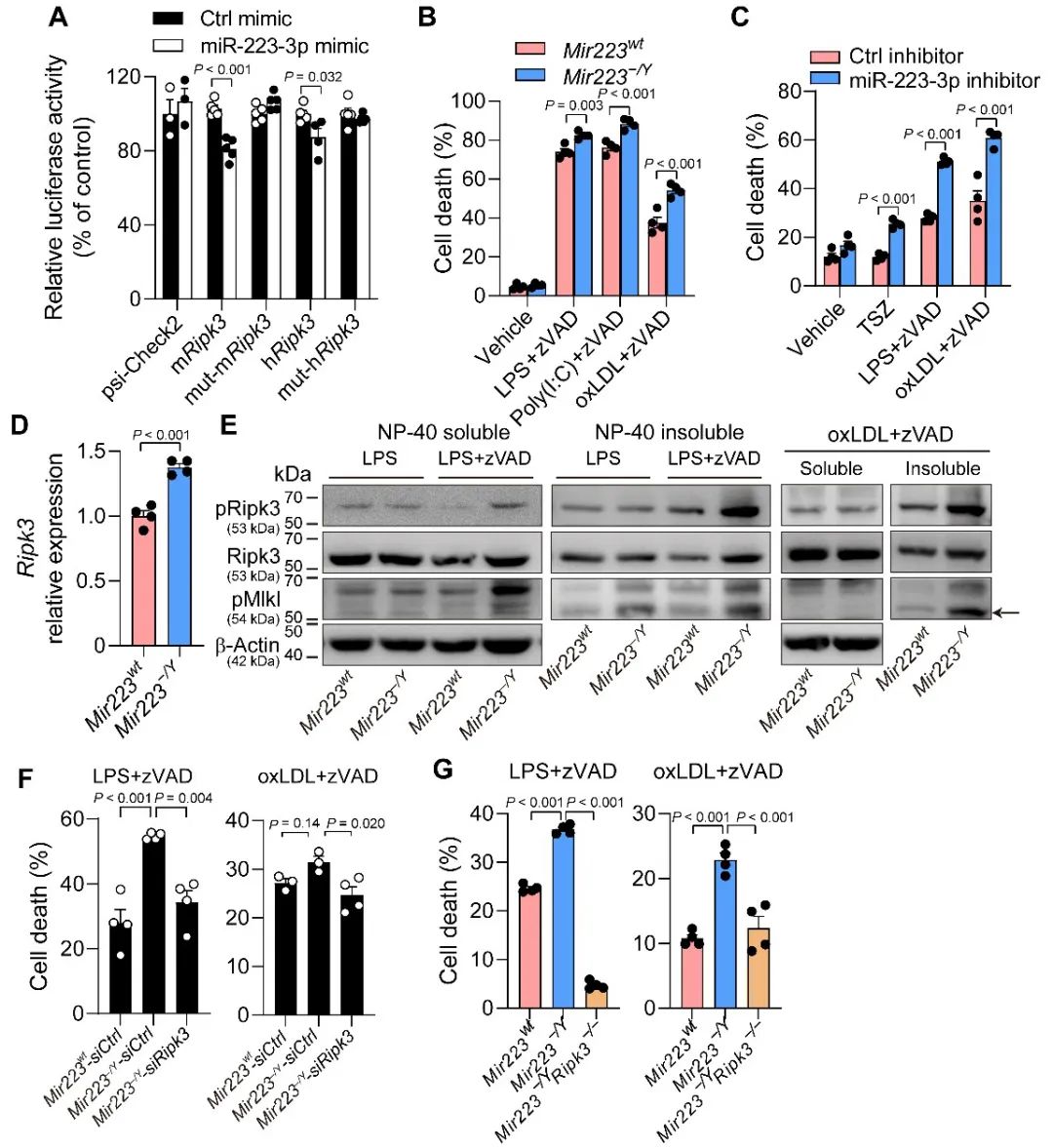
图1. miR-223-3p通过减少Ripk3的表达特异性抑制巨噬细胞的程序性坏死
为了探索巨噬细胞中的miR-223对动脉粥样硬化的影响以及确定该影响是否依赖于其对Ripk3的抑制作用,研究人员用骨髓移植的方法构建了Mir223wtApoe−/−、Mir223–/YApoe−/−和Mir223–/YRipk3–/–Apoe−/−骨髓嵌合小鼠并使用高胆固醇饲料喂食及左颈动脉结扎手术诱导的晚期动脉粥样硬化模型。骨髓细胞中Mir223基因的敲除加速了Apoe−/−小鼠的动脉粥样硬化发展,而缺失Ripk3或使用细胞程序性坏死抑制剂necostatin-1和GSK-872的处理可挽救Mir223缺失所带来的负面影响(图2)。与Mir223敲除一样,通过尾静脉注射的方法将miR-223-3p的抑制剂注入Apoe−/−小鼠的体内,显著增加了斑块面积,并促进了坏死核心的形成。
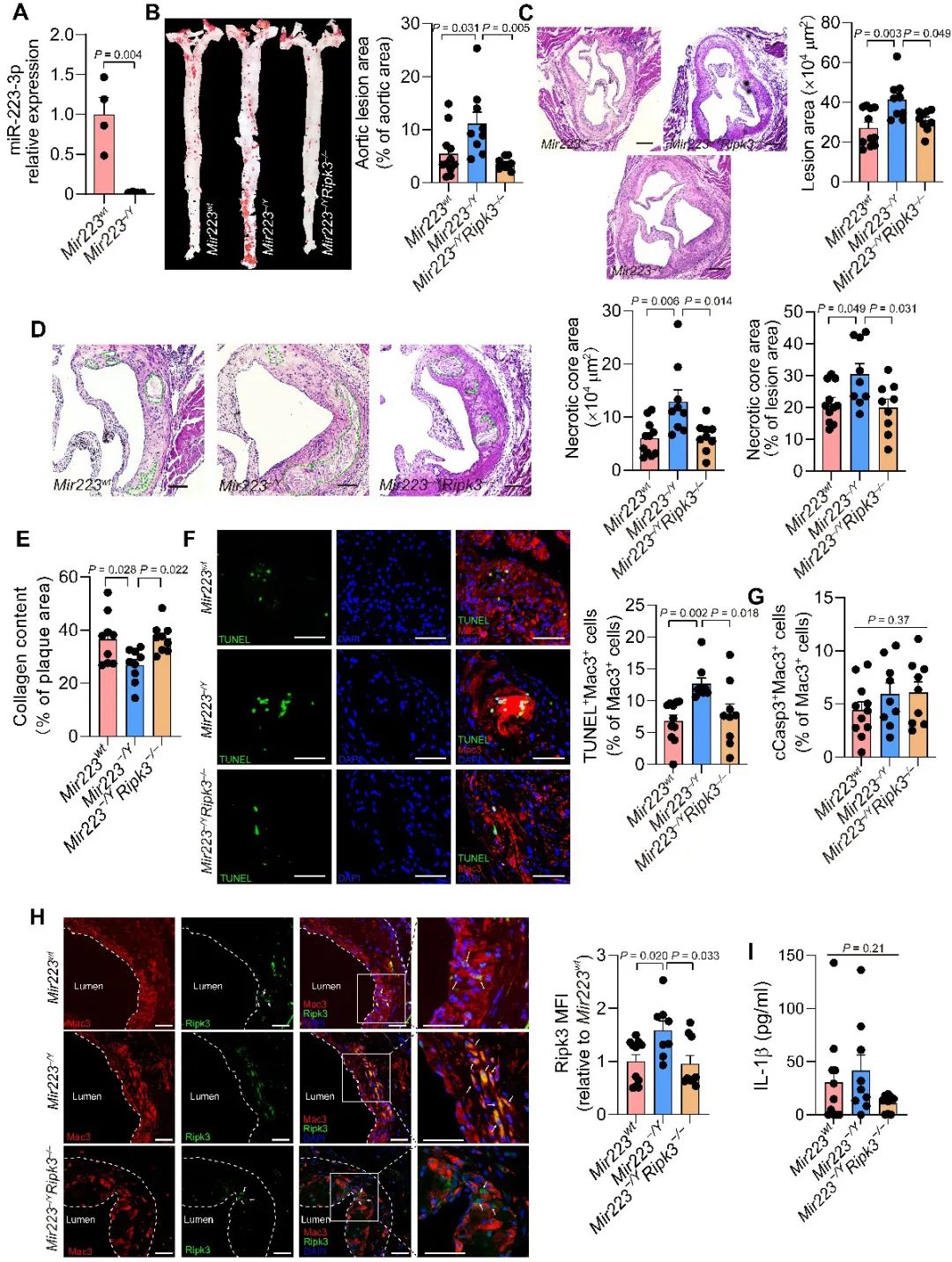
图2. 巨噬细胞中的miR-223通过抑制Ripk3减缓晚期动脉粥样硬化的发展
综上,程序性坏死在晚期动脉粥样硬化斑块中被显著激活,主要参与了坏死核心的形成,Ripk3/C/EBPβ/miR-223-3p形成的负反馈调控网络有助于泡沫巨噬细胞抵抗程序性坏死。miR-223-3p通过抑制巨噬细胞程序性坏死减缓了动脉粥样硬化的晚期发展(图3)。因此,miR-223-3p是一个具有治疗动脉粥样硬化潜在临床应用价值的分子。
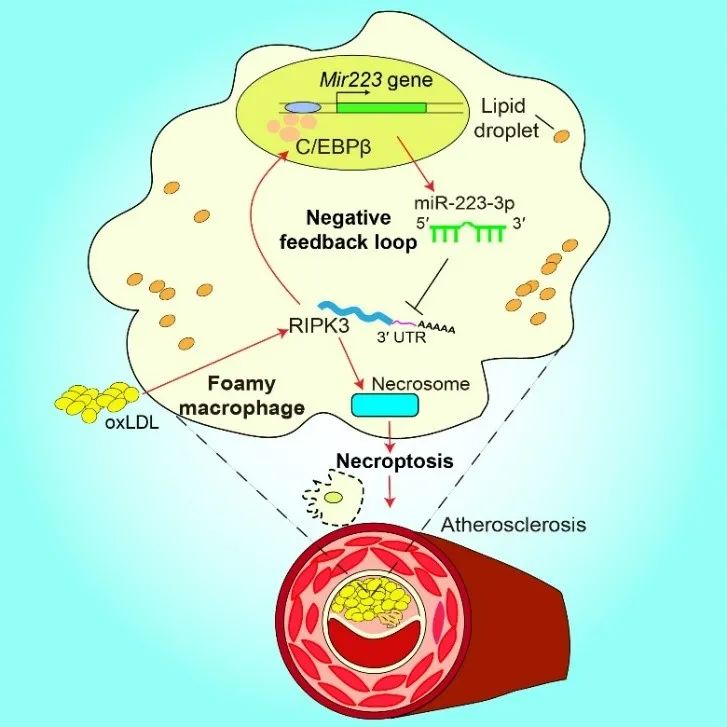
图3. 本文机制图
复旦大学基础医学院免疫学系博士生贾云慧和博士生程连平为论文的共同第一作者。复旦大学基础医学院免疫学系魏园园青年研究员和德国慕尼黑大学医院心血管病预防研究所Andreas Schober教授为共同通讯作者。研究获得了上海市科委面上项目(20ZR1406800)、国家自然科学基金(no.32000482)、海外高层次人才“青年项目”和上海高水平地方高校创新团队的项目资助。该研究的开展还得到了中山大学尹长军教授的指导。同时感谢复旦大学上海医学院公共技术服务平台和复旦大学基础医学院大型仪器共享平台的支持。
原文链接:
https://www.ahajournals.org/doi/abs/10.1161/ATVBAHA.123.319776
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言









#动脉粥样硬化# #RIPK3# #miR-223-3p#
37