
Nat Commun:席瑞斌/苗素生团队开发长读长转录组数据的分析工具包TAGET
2023-11-05 测序中国 测序中国 发表于上海

研究团队提出了一个名为TAGET的综合性计算工具包,用来分析Iso-seq全长转录组数据,包括转录本的映射和注释、基因融合的检测、差异表达分析以及差异同源异构体使用(DIU)分析。
基于第二代测序技术的RNA测序(RNA-seq)技术已被广泛用于各种转录组研究,但其测序的序列长度较短,大多只能覆盖转录本的一小部分,导致序列难以精确映射到同源异构体(isoform),使得RNA测序很难用于分析全长转录本。
长读长测序(Long-read sequencing)是Nature Methods评选出的2022年度技术,其主要优势是测序长度远大于二代测序平台。基于HiFi测序的PacBio RNA测序平台Iso-seq可以产生长而准确的序列。Iso-seq的准确性达到99.9%,N50读长约为2500bp,远长于平均转录本长度(约1300bp),为全长转录组分析提供了理想的技术平台。目前,科研人员已经开发了一些长读长测序数据的计算工具,但在剪切位点预测、转录本定量分析、基因融合探测等方面仍有很多计算问题需要解决,亟需发展更准确、系统的长读长转录组数据分析工具,以充分利用长读长测序技术的优势,推动转录组学研究的进步。
近日,北京大学数学科学学院的席瑞斌团队和哈尔滨医科大学附属肿瘤医院苗素生团队在Nature Communications上发表了题为“TAGET: A toolkit for analyzing full-length transcripts from long-read sequencing”的文章。研究团队提出了一个名为TAGET(Toolkit for Analyzing full-length GEne Transcripts)的综合性计算工具包,用来分析Iso-seq全长转录组数据,包括转录本的映射和注释、基因融合的检测、差异表达分析以及差异同源异构体使用(DIU)分析。通过实验验证和与RNA-seq数据的比较,研究团队发现TAGET比其它工具更准确地检测新剪切位点、同源异构体和基因融合。这项研究进一步完善了三代测序Iso-seq数据的分析流程,帮助研究人员更方便、更准确地进行全长转录组分析。

文章发表在Nature Communications
TAGET使用经过处理(polished)的转录本作为全长转录组分析的输入。根据Iso-seq数据的分析流程,TAGET只考虑由至少两个循环一致序列(CCS)支持的转录本。在映射阶段,TAGET整合了长读段映射方法和短读段映射方法的结果,并利用卷积神经网络(CNN)来提高剪切位点的预测准确性(图1b)。在得到映射结果后,TAGET将转录本注释为七大类,包括:FSM (Full Splice Match),ISM (Incomplete Splice Match),NIC (Novel in Catalog),NNC (Novel Not in Catalog),Genic,Intergenic和Fusion。随后,TAGET进行后续的基因融合检测和差异表达分析等(图1a)。
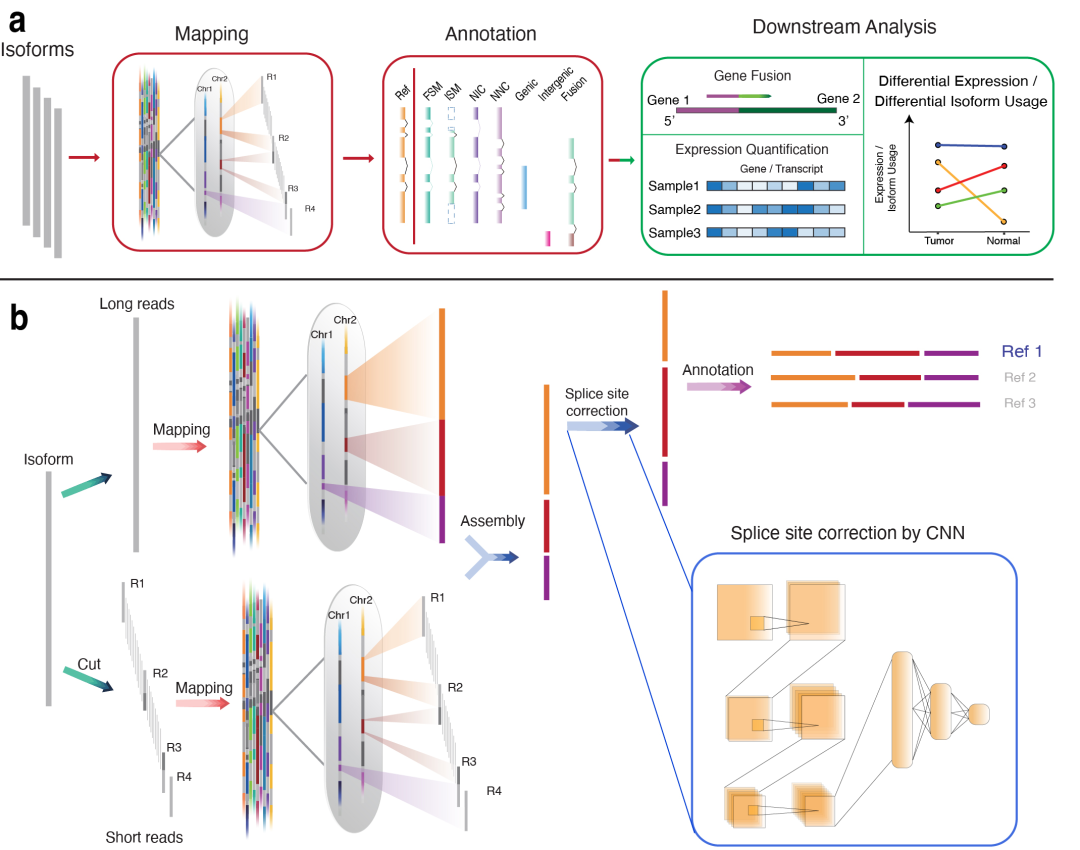
图1. TAGET工作流程
研究团队使用了来自公开的癌细胞系COLO829的数据集以及新测得的七对肿瘤和正常组织匹配的数据集来评估TAGET的算法表现。这七对新测得的数据中,有三对(LGC-133、LGC-415、LGC-265)来自喉癌(LGC)患者,另外三对(LUSC-07、LUSC-25、LUSC-39)来自肺鳞状细胞癌(LUSC)患者,还有一对来自骨肉瘤(OS)患者。对于OS患者的癌症和正常组织,分别进行了两次重复测序(OS-1和OS-2)。
研究团队首先比较了TAGET和长读段映射方法(minimap2、GMAP)的映射准确性。结果显示,TAGET预测的剪切位点相比minimap2和GMAP有更高的比例与Ensemble数据库中的剪切位点相匹配(图2a)。通过与RNA-seq数据的短序列数据和参考基因组进行比较,研究团队发现TAGET预测的剪切位点中有99.3%能够得到至少两个短读段或参考数据库的支持,略高于minimap2和GMAP(图2b)。对于Ensemble数据库中没有的新剪切位点,TAGET的预测结果中有75.2%能够得到至少两个短读段的支持,远高于 minimap2和GMAP的结果(图2c)。不使用CNN模型的TAGET预测的新剪切位点中大约有60%可以被RNA-seq支持(图2c),低于TAGET给出的预测结果,说明CNN模型的有效性(图2d)。
研究团队发现TAGET和SQANTI在转录本注释的结果上比较相似(图2e),其中注释差异较大的类别主要涉及Genic、FSM和ISM,这主要是因为TAGET和SQANTI对这些转录本类别的定义不同。在TAGET报告的新剪切位点中,有82.3%是典型的,远高于SQANTI(图2f)。Motif分析还证实了GT和AG分别在5'供体剪切位点和3'受体剪切位点上富集(图2g)。
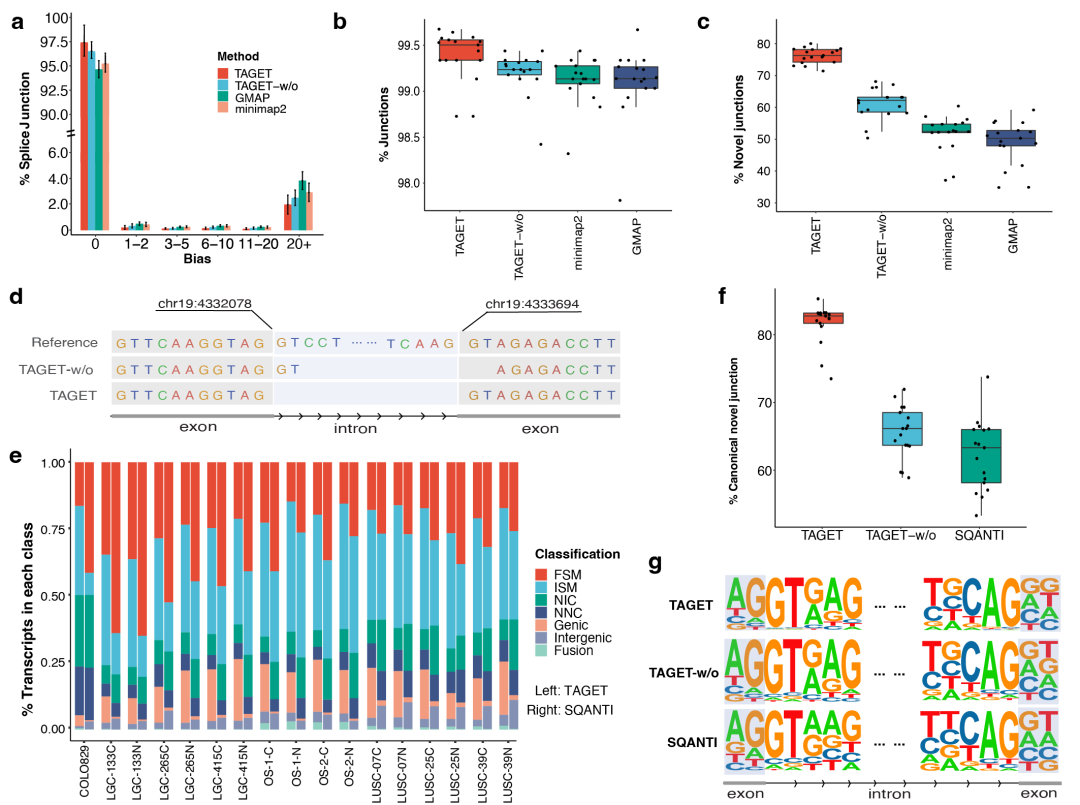
图2. TAGET映射和注释的性能评估
接下来,研究团队对TAGET的基因融合检测效果进行了分析,发现TAGET报告的基因融合比SQANTI和JAFFAL多(图3a),并且有更高比例的基因融合得到了至少2个RNA-seq序列的支持(图3b)。研究团队也通过模拟数据集证明了TAGET在灵敏度和精确度方面表现最佳(图3e)。
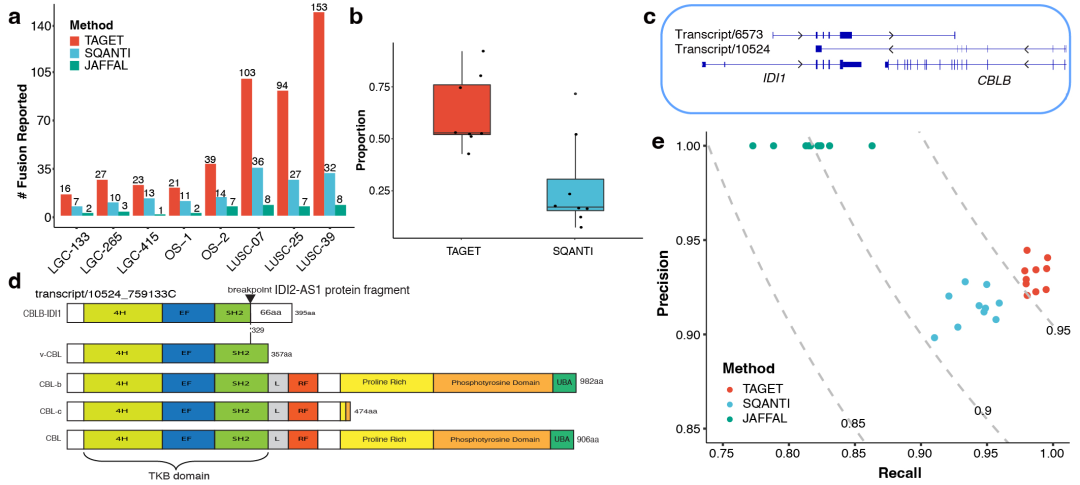
图3.检测基因融合
最后,研究团队使用GM12878细胞系的样本以及肺癌细胞系的23个样本来展示TAGET在牛津纳米孔技术(ONT)数据上的兼容性(图4)。
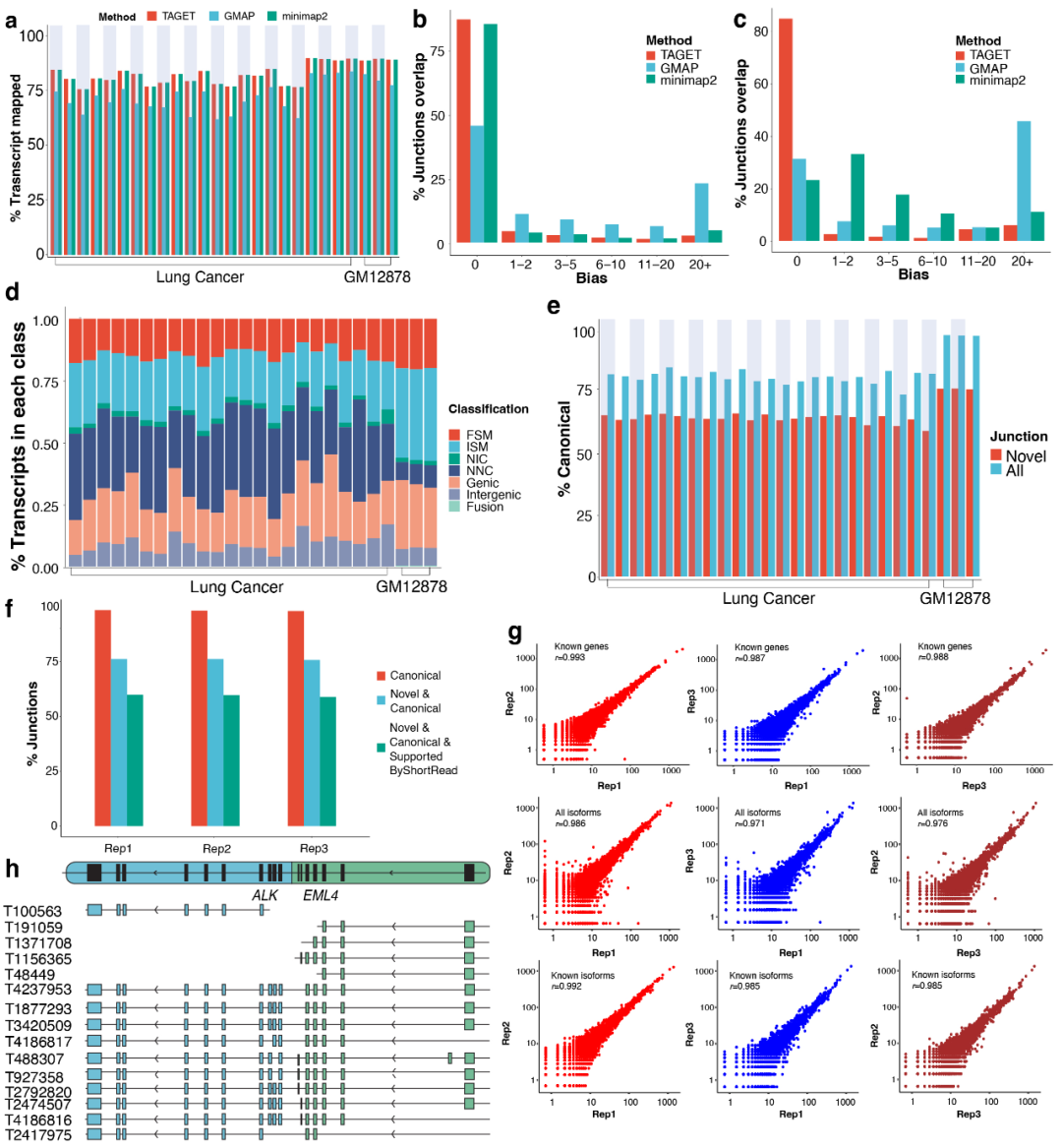
图4.TAGET在ONT数据集上的性能
综上所述,研究团队开发了一个可应用于全长转录组研究的多功能计算工具包TAGET,期望TAGET未来能在三代测序数据分析中发挥更重要的作用。
夏禹超、金子捷、张成胜、欧阳林坤是该研究的共同第一作者。该研究得到了国家重点研发计划、国家自然科学基金、黑龙江省博士后科研发展基金、北京信息科技大学基金、中国博士后科学基金、中俄数学中心的支持。
文章链接:
https://www.nature.com/articles/s41467-023-41649-0
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



