共价还是非共价?礼来首创新药BTK激酶抑制剂Pirtobrutinib启示
2023-02-08 精准药物 精准药物 发表于上海

近日, FDA 批准了礼来的Jaypirca:trade_mark:(吡妥布替尼Pirtobrutinib,100 mg 和50 mg 片剂),用于治疗复发或难治性套细胞淋巴瘤(MCL) 成年患者。
新年初始, FDA 批准了礼来的Jaypirca:trade_mark:(吡妥布替尼Pirtobrutinib,100 mg 和50 mg 片剂),用于治疗复发或难治性套细胞淋巴瘤(MCL) 成年患者,这些患者此前接受了至少两线全身治疗(包括布鲁顿氏酪氨酸激酶 (BTK) 抑制剂)。Jaypirca:trade_mark:通过FDA的加速批准途径获得批准,对该适应症的持续批准可能取决于验证性试验的结果。
Pirtobrutinib是第三代BTK抑制剂,也是第一款非共价型BTK抑制剂首创新药。

BTK激酶与致病性
布鲁顿氏酪氨酸激酶(BTK ,Bruton tyrosine kinase),是一种由人类BTK基因编码的酪氨酸激酶。这种酪氨酸激酶属于Tec激酶家族(在肝细胞癌中表达的酪氨酸激酶)。BTK激酶是一种包含5个信号结构域的659个氨基酸的蛋白质。在人类中,BTK蛋白质家族的成员主要在造血细胞中表达,尤其是在B细胞、骨髓细胞和血小板中。BTK在B细胞发育中起着至关重要的作用,它们的激活是抗原受体信号传导的第一步。
BTK基因突变与X 连锁无丙种球蛋白血症(XLA,X-linked agammaglobulinemia,亦称布鲁顿无丙种球蛋白血症)相关,这是一种原发性免疫缺陷病。已鉴定出至少400个BTK基因突变,其中至少有212种被认为是致病突变。[1]
BTK抑制剂
毫无例外地,所有BTK抑制剂药物的名称后缀都是tinib(替尼)。Tinib后缀是酪氨酸激酶抑制剂的标配名称: Tin代表酪氨酸激酶(tyrosin kinase),而ib则是抑制剂(inhibitor)的字母简写。
目前,BTK抑制剂主要应用于血液系统恶性肿瘤的治疗。BTK 也可能是治疗实体瘤的有希望的靶标。目前上市的BTK抑制剂包括:
-
Ibrutinib依鲁替尼:套细胞淋巴瘤、慢性淋巴细胞白血病和华氏巨球蛋白血症
-
Acalabrutinib阿卡拉布替尼:复发性套细胞淋巴瘤
-
Zanubrutinib赞布替尼:套细胞淋巴瘤
-
Tirabrutinib替拉布替尼:复发性或难治性原发性中枢神经系统淋巴瘤,在日本上市
-
Pirtobrutinib吡布替尼:复发性 B 细胞淋巴瘤或 B 细胞白血病
-
Orelabrutinib奥布替尼:套细胞淋巴瘤或慢性淋巴细胞白血病/小淋巴细胞淋巴瘤,在中国上市
目前处在III期临床的BTK抑制剂候选药物包括:
-
Acalabrutinib:复发性慢性淋巴细胞白血病 (CLL),据报道总体缓解率为 95%。
-
Evobrutinib:多发性硬化症
-
Tolebrutinib:多发性硬化症
-
Remibrutinib:多发性硬化症
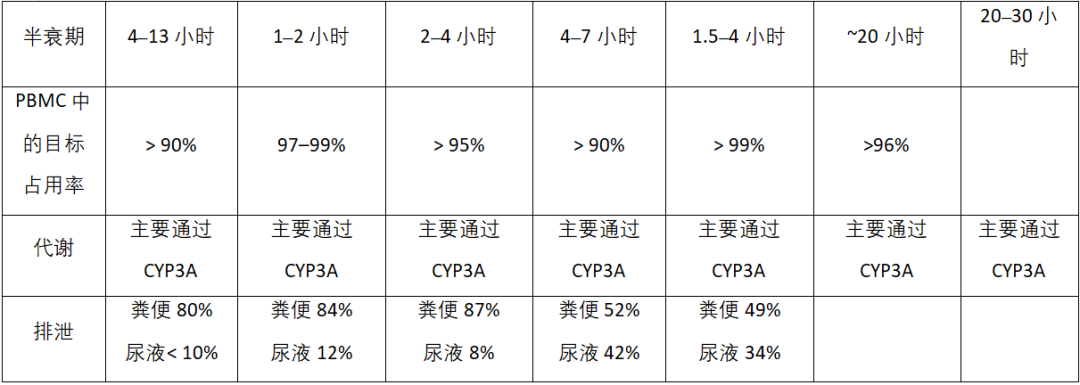
Pirtobrutinib是Lilly开发的一款BTK高选择性抑制剂药物,用于治疗复发或难治性套细胞淋巴瘤。[2] 尽管BTK抑制剂的同类药物中还有多种其他药物,但每种药物都具有不同的特性和药理特性。[3]上市以及部分在研(Nemtabrutinib在研;Orelabrutinib在中国上市;Tirabrutinib在日本上市)BTK抑制剂属性总结见表1。[3]
表1. 上市以及部分在研BTK抑制剂药物属性
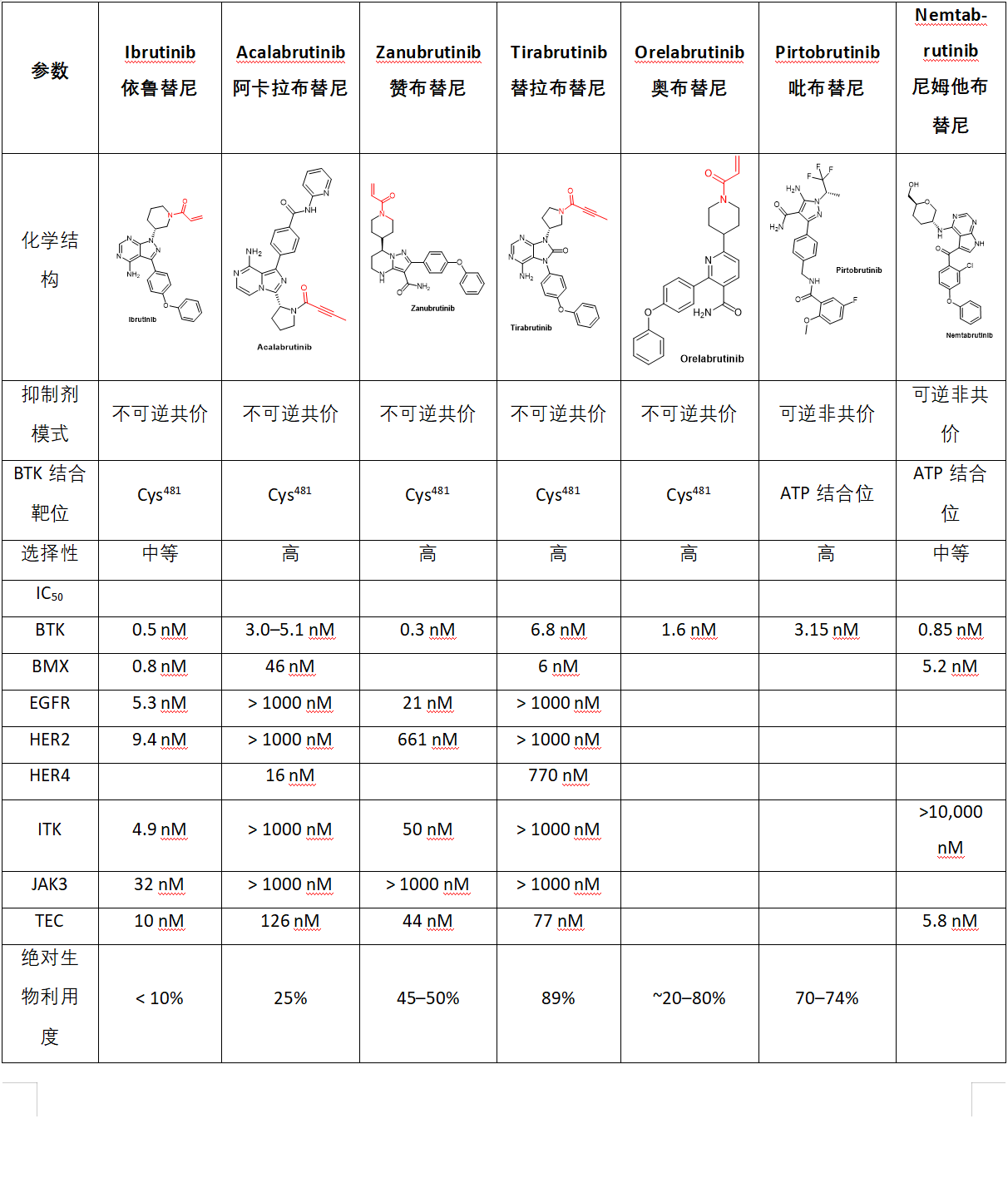
我们先以第一代BTK抑制剂Ibrutinib为例,它是一种有效的、不可逆的布鲁顿酪氨酸激酶(BTK)抑制剂。因为BTK在多种B细胞恶性肿瘤的发病机制中发挥作用,包括套细胞淋巴瘤(MCL)、弥漫性大B细胞淋巴瘤(DLBCL)、 滤泡性淋巴瘤和慢性淋巴细胞白血病(CLL),因此抑制BTK的生物活性是治疗这些适应症的重要手段。Ibrutinib可有效抑制BTK,从而抑制B细胞受体通路,该通路在B细胞癌中通常异常活跃。抑制BTK可以限制降低体内恶性B细胞增殖和存活。
共价抑制剂与非共价抑制剂
共价与非共价抑制剂概念
值得注意的是,Ibrutinib是一款可逆共价药物。所谓的共价药物,指的是药物分子含有活性的“弹头”(warhead)基团,通常是亲电基团,例如丙烯酰胺、环氧乙烷、有机硼酸、氰基、羰基、乙烯磺酰胺、β-内酰胺、α-酮酰胺等。这些药物分子上的亲电基团可以与受体上广泛存在的亲核基团,诸如巯基、羟基、氨基和咪唑基等发生亲电亲核反应,产生共价键,从而更高地结合受体。
而传统的药物分子,绝大多数都是非共价药物,它们与受体之间的结合,通常是利用与结合口袋之间的非共价作用,例如氢键、离子键、偶极-偶极作用,疏水作用,芳香环作用等。尽管非共价药物在比例上占据绝大多数,但它的作用方式是一个可逆过程,即结合与解吸附之间的平衡过程。与受体结合的非共价药物分子,在改变的环境中可以从受体-药物加合物上解吸附下来。
因此要实现对致病过程的有效控制,就需要较高的药物暴露量,因此要求更大的药物剂量,使得结合-解析附的这个平衡过程朝向结合的方向运动,从而实现对靶向受体的有效抑制。但这种策略导致了剂量大、药效时间短、给药频率高的问题。正是非共价药物的这些限制,催化了共价药物的开发过程。
虽然共价键的形成与裂解也可能是一个平衡过程(针对可逆共价药物),但相对于非共价键来说,一旦产生新的共价键,体系中的低能量是不足以将其裂解的,因而药物-底物共价加合物可以相对稳定地存在。
除了这些共价药物的内在优势之外,对于那些结合位点不强烈的所谓“不可成药”(undruggable)受体来说,寻找合适的非共价结合口袋可能是一件缘木求鱼的努力。在这种情况下,开发共价药物或许是靶向这些“不可成药受体”更好的选择。例如undruggable受体KRASG12C 蛋白的抑制剂sotorasib(图1,红色丙烯酰胺结构为共价药物弹头),为治疗非小细胞肺癌提供了方案(以sotorasib为原料药的Lumakras 和 Lumykras已经上市)。
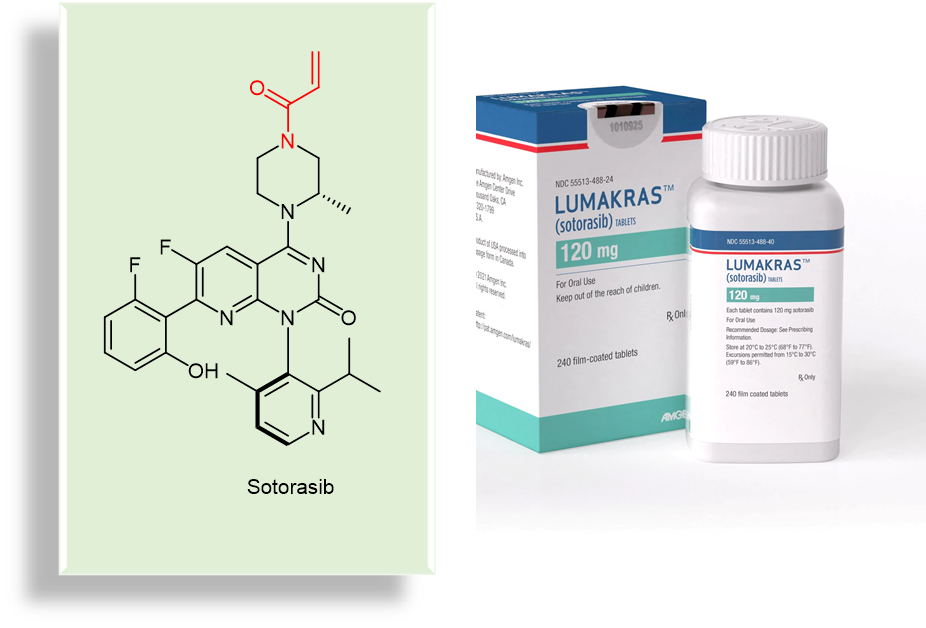
图1. Sotorasib化学结构(红色部分为丙烯酰胺弹头)
虽然共价药物属于药物发展史上的创新类药物,但人类对于共价药物的利用,可以追溯到1899年的阿司匹林,它的乙酸芳香酯基团(乙酰水杨酸)赋予了它共价药物的身份。其消炎抗凝血的本质机理,就是通过其分子中的乙酰基团从药物分子转移到受体蛋白酶的丝氨酸侧链实现的(图2)。
1928年问世的抗生素青霉素,也是利用它的共价药物身份拯救生灵无数的。它的分子中包含了标志性的β-内酰胺四元环,同样具有高度的反应活性,可以与很多亲核发生开环反应,从而实现对于靶向受体的抑制作用。
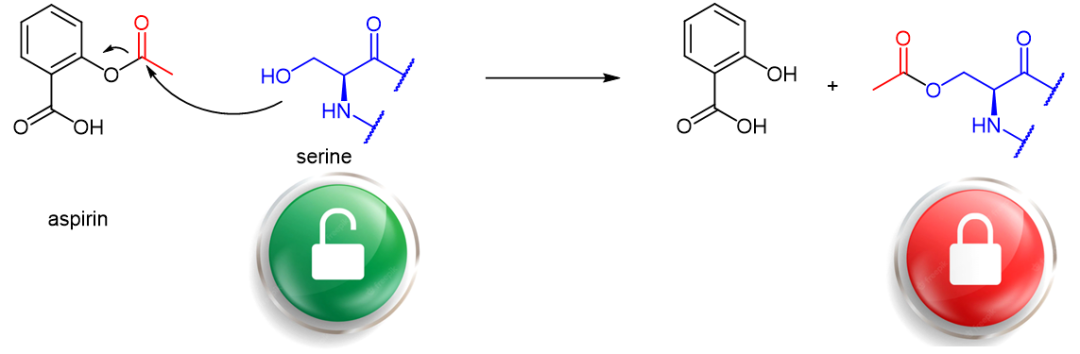
图2. 阿司匹林抑制丝氨酸蛋白酶功能的化学机理
在人类药物的发展历史上,Ibrutinib可以说是一个具有里程碑意义的共价药物开发。虽然它之前有过几款含有亚砜(奥美拉唑)、环氧乙烷(磷霉素)、有机硼酸(万珂)和氰基(沙格列汀)的共价药物(图3),但是Ibrutinib的成功上市催化了大量共价药物的涌现,而且多数的共价药物同Ibrutinib一样,采用了丙烯酰胺的弹头结构作为共价药物的核心构件。

图3. 共价键药物发展历史图(图片来源:E药学苑)
可逆共价抑制剂Nirmatrelvir
在抗SARS-CoV-2病毒的战争中发挥关键作用的Paxlovid,它的有效成分nirmatrelvir也是一款共价药物,它的亲电弹头为氰基。氰基是一个相对比较温和的亲电基团,因此也增强了nirmatrelvir的选择性,降低了脱靶效应。
Nirmatrelvir靶向的SARS-CoV-2病毒的3C样蛋白酶(3C-like protease, 亦称main protease,简称为3CLPro或MPro),属于半胱氨酸蛋白酶家族。Nirmatrelvir上的氰基基团在其与MPro非共价结合之后,亲电进攻MPro受体上的Cys145残基的侧链巯基,产生硫代亚胺酸酯(thioimidate)加合物中间体(图4)。
因为这个过程形成了新的共价键,因此是一个共价反应过程,nirmatrelvir被视为共价药物。之所以将nirmatrelvir的共价药物前面冠以“可逆”的标签,是因为硫代亚胺酸酯thioimidate并不是可以完全稳定存在的最终产物,它可以被水解为酰胺和硫醇化合物(图4)。这个过程重新释放了硫醇底物Cys145,因此被视为可逆过程(致病的MPro被重新释放)。
不过从有机化学的角度来看,这并不是一个可逆过程,因为它只是再生了一个反应物硫醇,但并没有实现彻底的可逆,因为氰基化合物没有再生。它只是一个医学上的可逆过程,但不是有机化学的可逆反应。不管如何,在医学界,像nirmatrelvir这样的抑制剂被看作可逆共价抑制剂。
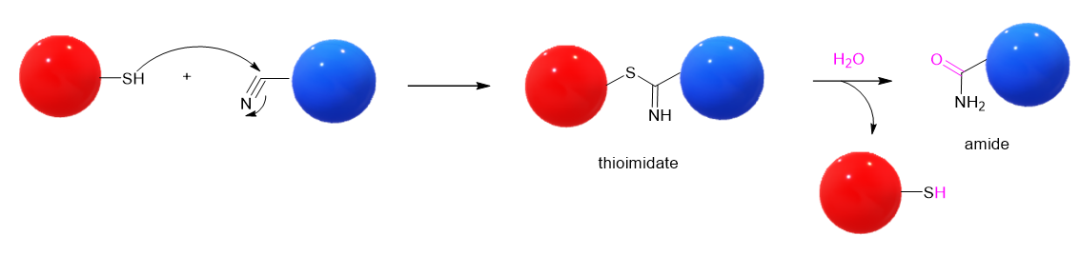
图4. 巯基化合物与腈类化合物产生硫代亚胺酸酯,以及硫代亚胺酸酯水解的反应
不可逆共价抑制剂adagrasib

去年年底刚得到FDA批准上市的adagrasib(Krazati:registered:)是一款不可逆共价抑制剂,用于患有KRASG12C突变的转移性非小细胞肺癌的成人患者。[4] 它的靶标是KRASG12C突变蛋白(KRAS的第12位氨基酸残基由甘氨酸Gly突变为半胱氨酸Cys)。KRAS抑制剂的开发难点上文提及,在于KRASG12C缺乏明确的结合口袋,具有一定的undruggable(不可成药性)属性。[5] 基于这一考虑,开发人员转向了共价药物的思路。

图5. Adagrasib化学结构
通过亲电弹头的引入,实现药物分子与靶标之间的共价作用,从而规避了不可成药性的限制。通过观察adagrasib的化学结构,人们就不难发现两个“卓尔不群”的基团:2-氟丙烯酰胺与氰基。理论上这两个亲电基团都可以当作弹头使用,它们同时出现在同一分子内并不多见。
在adagrasib中,真正起到共价弹头作用的是2-氟丙烯酰胺结构。它的亲电性比氰基更强,靶向的正是KRASG12C突变蛋白中致病的突变12位半胱氨酸。2-氟丙烯酰胺与半胱氨酸巯基之间的反应,从有机合成的角度来看属于迈克尔加成反应(Michael-addition),迈克尔供体为巯基,受体为2-氟丙烯酰胺。
它们之间的反应产生了新的化学键,因此adagrasib是共价药物。新生成的化合物Adagrasib-Cys加合物含有2-氟-3-硫醚结构。和上文提到的nirmatrelvir的硫代亚胺酸酯不同,2-氟-3-硫醚是个相当稳定的结构,被共价结合的Cys12很难实现重生,有点“咬定青山不放松”的意味。出于2-氟-3-硫醚的高度稳定性,这个过程被视为不可逆的结合,因此人们称呼adagrasib为不可逆共价抑制剂。
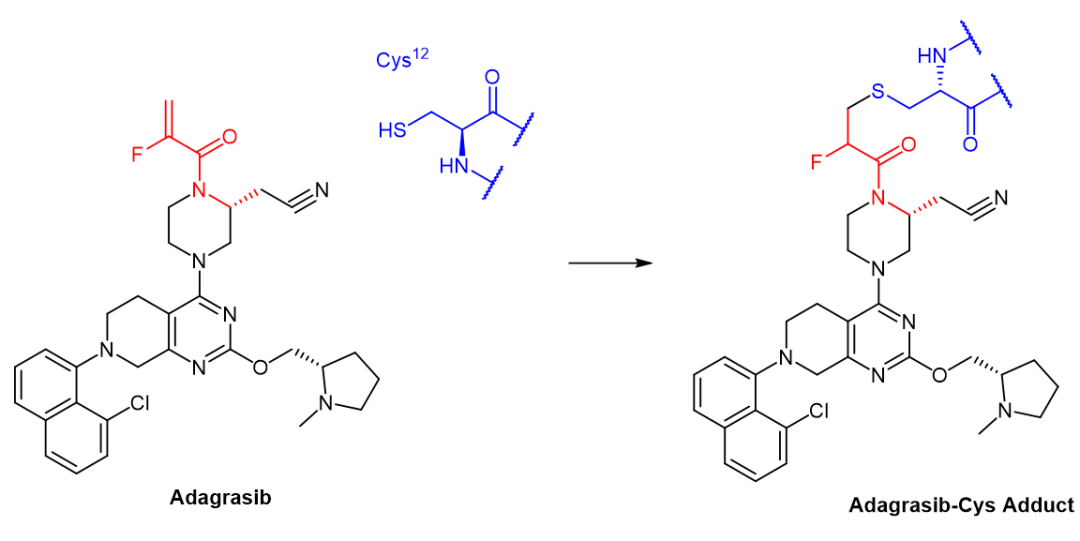
图6. Adagrasib共价不可逆结合KRASG12C的化学反应
BTK共价抑制剂
表1中总结的BTK抑制剂中,在Pirtobrutinib之前上市的无一例外均是共价抑制剂。图7显示了它们的化学结构,从中可以发现,它们均包含丙烯酰胺或者炔基酰胺这样的亲电弹头结构。从抑制BTK的化学反应角度来看,它们都是不可逆共价抑制剂。
例如Ibrutinib,它与BTK活性位点中的半胱氨酸残基(Cys481)形成共价键,从而导致其抑制。BTK的抑制在B细胞受体信号传导中发挥作用,因此,Ibrutinib的存在可防止下游底物(如PLC-γ)的磷酸化。[6]
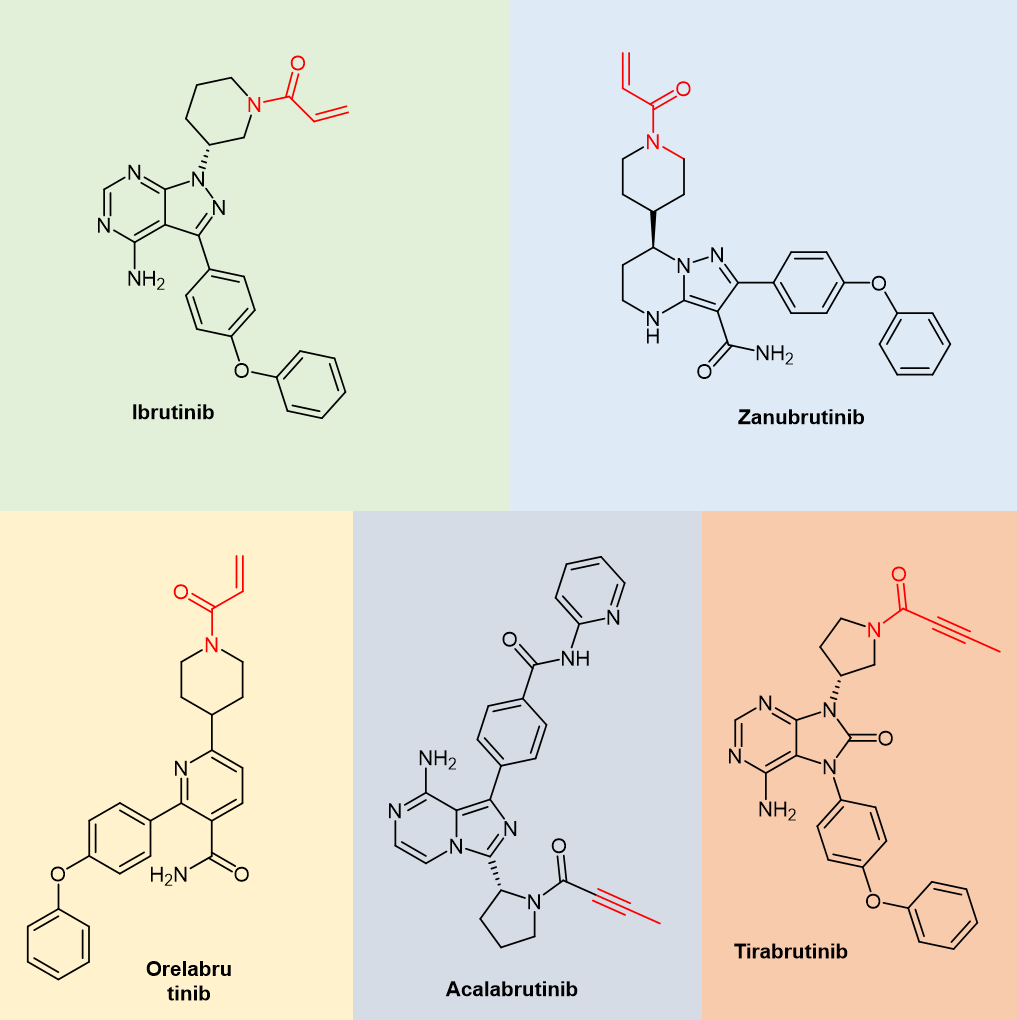
图7. 上市BTK共价抑制化学结构(红色部分为亲电弹头)
让我们再近距离观察一下第二代BTK抑制剂代表acalabrutinib。Acalabrutinib 及其活性代谢物 ACP-5862(图8)与BTK活性位点的半胱氨酸残基 (Cys481) 形成共价键,从而抑制BTK酶活性。[7] 因此,acalabrutinib抑制BTK介导的下游信号蛋白CD86和CD69的激活,最终抑制恶性B细胞增殖和存活。[8]

图8. Acalabrutinib与其代谢物ACP-5862化学结构
尽管Ibrutinib通常被认为是首创新药的BTK抑制剂,但Acalabrutinib被认为是第二代BTK抑制剂(zanubrutinib, tirabrutinib和orelabrutinib也被视为第二代BTK抑制剂),主要是因为它表现出更高的选择性和对 BTK 靶向活性的抑制,同时具有更高的IC50 ,以及几乎不抑制ITK、EGFR、ERBB2、ERBB4、JAK3、BLK、FGR、FYN、HCK、LCK、LYN、SRC和YES1.2的激酶活性。
实际上,acalabrutinib 被合理地设计为比 ibrutinib 更有效和更具选择性,同时在理论上表现出更少的副作用,因为该药物的脱靶效应最小。
BTK抑制剂第三代:非共价抑制剂Pirtobrutinib
刚得到FDA批准的Pirtobrutinib是BTK抑制剂的第三代,属于传统的非共价药物。虽然身份与绝大多数的小分子药物一样,但对于BTK抑制剂家族来说,却是一个创新的类型,因为之前上市的BTK抑制剂无一例外地都属于不可逆共价药物。为什么回归传统却实现了创新的效果?这就涉及到共价药物不可回避的缺陷了。
虽然共价键药物有着药效时间长和剂量低的优势,但是活性过高的亲电弹头是否会无差别地攻击体内众多的亲核基团,这是药物开发者高度关注的内容。共价药物潜在的脱靶效应高风险,以及可能引发的免疫响应,这些涉及药物安全性问题的隐患,都是共价药物潜在的“阿喀琉斯之踵”。
对于BTK抑制剂而言,靶向Cys481的策略的负面效应是,你必须依赖这个关键的半胱氨酸。就像足球比赛中,教练部署重点打击某条边路,一旦对方看破了这个招数,在你重点打击的位置换上来一个狠人的话,球队的进攻有可能顿时失去方向。
致病与治病也一样,生物体对于不断给予的药物,可能会以耐药性(drug resistance)的方式负隅反抗。共价BTK抑制剂,无论是第一代的Ibrutinib,还是第二代的Acalabrutinib,靶向的都是BTK活性位点的Cys481,通过与半胱氨酸侧链的巯基形成共价结合物的方式抑制BTK酶活性。
而BTK采取的耐药性对策,正是通过对关键的Cys481残基变异而实现的,例如Cys被Ser取代而产生的BTKC481S(尽管丝氨酸侧链的羟基也是一个亲核基团,但相对于半胱氨酸侧链的巯基的亲核性就弱了很多)[9]。
当BTK抑制剂中的亲电性弹头无法寻找到靶点上的半胱氨酸巯基时,这款药物的效应就大打折扣了。人们开始越来越关注对共价BTK抑制剂产生的耐药性,而耐药性也导致了大量患者停止治疗的严重后果。
如表1所示,共价BTK抑制剂的口服生物利用度较低,半衰期较短。这可能导致BTK靶点覆盖率在快速增殖的肿瘤中不理想,BTK蛋白周转率高,最终在一些患者中表现为获得性耐药。
为了解决这些局限性,研究人员开发了一种高度选择性的非共价 BTK抑制剂Pirtobrutinib。1/2期BRUIN研究的初步结果表明,无论既往治疗方式、既往治疗线数或BTK的C481突变状态如何,Pirtobrutinib在CLL/SLL患者中均具有良好的耐受性并显示出良好的疗效。
Pirtobrutinib是第一个非共价可逆(其实非共价就已经可以代表可逆了,不存在非共价不可逆结合)结合特性的药物。这可以实现更高的选择性,减少脱靶效应的不利影响。Pirtobrutinib因此具有治疗复发性或复发性CLL的能力。
作为第三代BTK抑制剂,Pirtobrutinib不依赖于与活性位点的Cys481结合,因此不会产生明显耐药性。得益于显著延长的半衰期,当身体合成新的BTK 时,Pirtobrutinib将留在体内并引起持续抑制。这与那些半衰期过短的共价抑制剂所导致的抑制间隙形成了明显的对比。
— 总结—
Pirtobrutinib的批准为复发性或难治性MCL (套细胞淋巴瘤) 患者带来了一个重要福音,他们目前的选择有限,并且在停止使用共价BTK抑制剂治疗后会导致预后不良。Pirtobrutinib可以为以前接受过共价BTK抑制剂治疗的患者提供疗效,有可能延长患者的受益时间。
Pirtobrutinib提供了一种新方法,可以在使用共价BTK抑制剂治疗后,靶向BTK通路,并且对复发和难治性MCL (套细胞淋巴瘤) 患者的治疗模式产生有意义影响的潜力。
这篇文章阐述了共价药物的产生与发展案例,并使用BTK抑制剂作为媒介阐述了共价药物起效的机理,特点,以及可能存在的问题。需要注意的是,对于非共价药物与共价药物,我们不应该采用非此即彼的逻辑来割裂看待。事实上,两类药物都有极大的可取之处,否则也不会存在这么多的成功案例。
实际上,共价药物的发展,是在非共价药物的坚实基础之上实现的,否则随便合成一些具有亲电性的化合物就可以充当药物靶向受体了?很多成功的共价药物,是在对受体结合充分的了解基础之上,在有效的非共价药物的恰当位置,加入合理的亲电弹头实现的。这是很多成功的共价药物的设计策略,包括Paxlovid中的拟肽nirmatrelvir。
在非共价的基础之上实现共价,仍将是未来开发共价药物的重要途径之一。而共价药物引发的耐药性、选择性、安全性等问题,又需要非共价药物去“待从头,收拾旧山河”。两类药物的互相借鉴、促进与弥补,正是药物设计不断进步的动力之一。
参考文献(上下滑动查看更多)
[1] Šimčíková, D. et al. Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases. Scientific Reports. 2019, 9, 18577.
[2] BTK Inhibitor Molecule Overview | Loxo Oncology. Lillyloxooncologypipeline.com.
https://www.lillyloxooncologypipeline.com/molecule/btk-inhibitor?gclid=CjwKCAjwv-GUBhAzEiwASUMm4rr69kaDRjX_VR-
HBgVo6TGVW2OwrVw3S1Nvuh9EXFoB6Jjd87XUnhoCh5QQAvD_BwE.
[3] Shirley, M. Bruton Tyrosine Kinase Inhibitors in B-Cell Malignancies: Their Use and Differential Features. Target Oncol. 2022, 17, 69-84.
[4] KRAZATI:registered: (adagrasib), FDA Approved for Treatment of Advanced Non-Small Cell Lung Cancer Harboring the KRASG12C Mutation, Available at Biologics by McKesson. MeKesson Announcement. 14, 12, 2022.
[5] Christensen, J. G. et al. The KRASG12C Inhibitor, MRTX849, Provides Insight Toward Therapeutic Susceptibility of KRAS Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2019, pii: 2159-8290.CD-19-1167.
[6] Davids, M. S. et al. Ibrutinib: a first in class covalent inhibitor of Bruton's tyrosine kinase. Future Oncol. 2014, 10, 957-967.
[7] Cheah, C. Y. et al. Mantle Cell Lymphoma. J Clin Oncol. 2016, 34, 1256-1269.
[8] Lymphoma Research Foundation's Getting the Facts: Mantle Cell Lymphoma - Relapsed/Refractory. https://lymphoma.org/wp-content/uploads/2018/04/MCL-Relapsed_Refractory.pdf
[9] Woyach, J. A. et al. Efficacy and Safety of Nemtabrutinib, a Wild-Type and C481S-Mutated Bruton Tyrosine Kinase Inhibitor for B-Cell Malignancies: Updated Analysis of the Open-Label Phase 1/2 Dose-Expansion Bellwave-001 Study. Blood. 2022, 140 (Supplement 1), 7004–7006.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




