
Cell:TR-gnomAD:揭示遗传多样性中串联重复的全新视角
2024-04-13 生物探索 生物探索 发表于陕西省

TR-gnomAD的成果不仅提供了一个宝贵的科研资源,也强调了在全球多样性背景下研究人类基因组变异的重要性。
引言
在人类基因组的研究中,串联重复(Tandem Repeat, TR)的扩展是一个重要且复杂的领域。串联重复是指基因组中连续重复的DNA序列,它们在进化和疾病形成中发挥着关键作用。尽管它们构成了我们基因组的约6%,但到目前为止,全球性的生物样本库(biobank-scale)对这类遗传变异的参考图谱仍然非常有限。基因组聚合数据库(Genome Aggregation Database, gnomAD)虽然被广泛认为是单核苷酸变异(Single-Nucleotide Variants, SNVs)和结构变异(Structural Variants, SVs)的黄金标准参考图谱,但对于串联重复扩展的研究却相对缺乏。
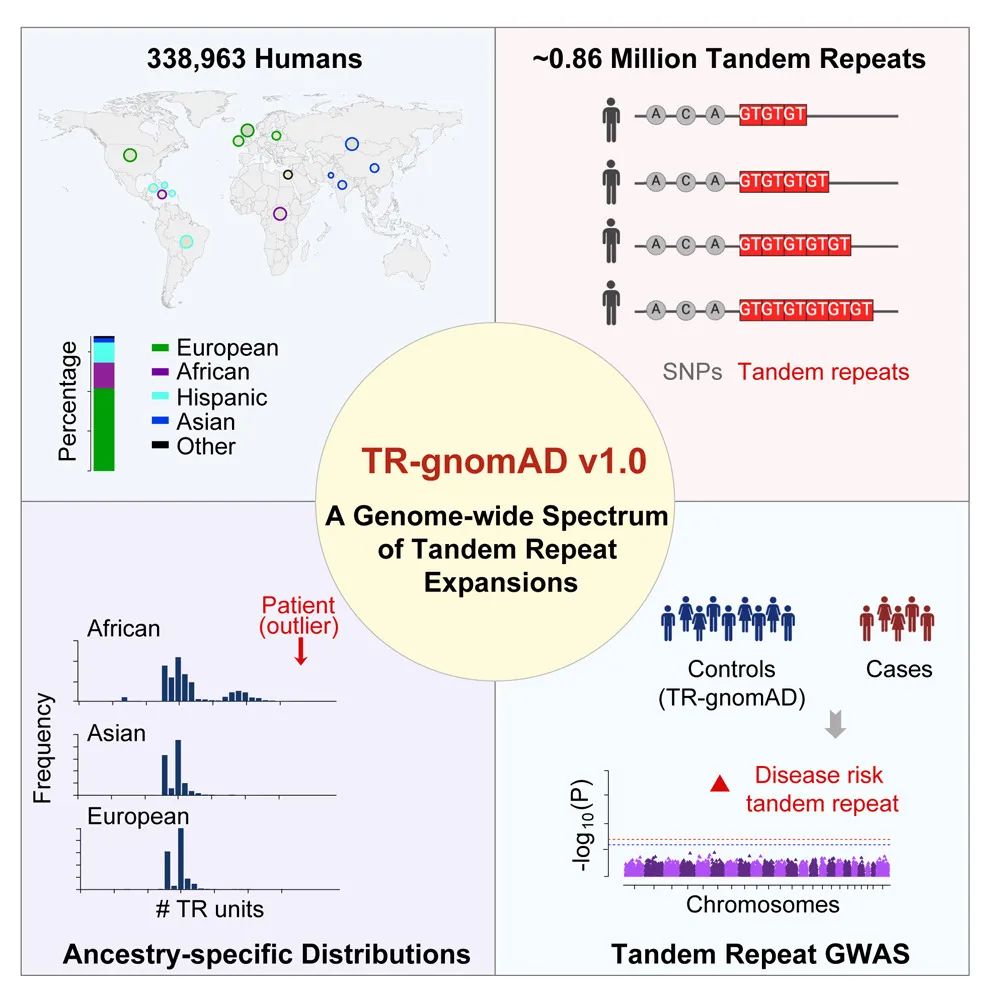
为了弥补这一空白,加州大学欧文分校(University of California, Irvine, UCI)的研究团队发起了TR-gnomAD项目。这个项目创建了一个涵盖338,963个人类全基因组测序(Whole-Genome Sequencing, WGS)样本的串联重复扩展的参考图谱,特别关注包含大量非欧洲血统样本(占39.5%)的多样性。TR-gnomAD不仅提供了一个关于串联重复单元数量频率在不同人种间差异的重要视角,而且还揭示了特定串联重复扩展在疾病群体中的潜在致病性。
这项工作的重要性在于其对疾病相关串联重复的识别和解释能力,为罕见病(rare diseases)诊断和治疗提供了新的策略和工具。TR-gnomAD通过使用ExpansionHunter和GangSTR这两种精确的串联重复基因型分析工具,能够在大规模样本中高效地识别和分类这些遗传标记。此外,该项目还开发了基于2-Wasserstein距离的串联重复差异分数(Tandem Repeat Disparity Score, TRDS),这是一个数学工具,用于量化不同人群间串联重复单元数量分布的差异。
通过这一全新的参考图谱,研究人员和临床医生现在可以更准确地评估个体中特定串联重复的扩展与疾病之间的联系,从而促进精准医学(Precision Medicine)的发展。TR-gnomAD的成果不仅提供了一个宝贵的科研资源,也强调了在全球多样性背景下研究人类基因组变异的重要性。(4月5日 Cell “A genome-wide spectrum of tandem repeat expansions in 338,963 humans”)

Highlights
大规模Biobank参考图谱:该研究创建了一个包含338,963个全基因组测序样本的串联重复(tandem repeats, TR)扩展的参考图谱。这是一个涵盖多种祖先背景(其中39.5%为非欧洲样本)的大规模数据集,为研究串联重复扩展提供了宝贵的资源。
多祖先背景的串联重复参考地图:该研究不仅提供了关于TR在不同人群中的分布数据,还能揭示特定祖先群体中TR扩展的疾病相关性。通过比较不同祖先群体间的TR单元数量频率差异,研究揭示了特定的TR扩展与疾病间的关联。
区分良性与潜在病理性TR扩展:利用生物信息学工具,TR-gnomAD能够区分在人群中常见的可能良性的TR扩展和那些在疾病群体中更频繁出现的潜在病理性TR扩展。这对于临床上解释TR扩展在遗传病中的意义极为重要。
高质量的TR分型技术:通过使用两种准确的TR分型工具——ExpansionHunter和GangSTR——该研究提高了TR分型的覆盖率和准确性。这使得研究者能更好地分析TR的多样性及其与疾病的关联。
(Credit:Cell)
Strategies
该研究构建了一个名为TR-gnomAD的基因库规模的串联重复扩展(tandem repeat expansions, TRs)参考图谱,涵盖了338,963名人类基因组样本。这些样本代表了多样的血统,其中39.5%来自非欧洲样本。研究中使用了两种准确且广泛使用的串联重复基因分型工具,ExpansionHunter和GangSTR,以提高串联重复基因型的覆盖率。
首先,通过ExpansionHunter和GangSTR对每个样本中的串联重复进行基因分型。这些工具能够准确识别并计数串联重复单元(TR units)。然后,使用TRTools中的MergeSTR和dumpSTR等工具对基因型数据进行整合和质量控制,以确保数据的可靠性。
随后,研究团队开发了一种基于2-Wasserstein distance的串联重复差异评分(TR disparity score, TRDS),用于量化不同群体间串联重复单元数频率分布的差异。此评分帮助研究者探究特定血统中串联重复扩展的流行病学特征。
此外,TR-gnomAD还可以作为控制队列,用于解释已知的临床病理性串联重复。通过与疾病组中的串联重复单元数进行比较,研究人员可以识别出潜在的病理性扩展。
总体而言,TR-gnomAD提供了一个宝贵的资源,可用于研究和诊断与串联重复扩展相关的遗传疾病,特别是在多种血统中。这项资源的开放获取性和高覆盖率使其成为解读人类遗传多样性中串联重复的重要工具。
Behind the Scenes
提高种群代表性
作者强调了在未来研究中,将增加更多种群的全基因组测序数据(Whole-Genome Sequencing, WGS),尤其是那些在现有研究中代表性不足的种群。这表明TR-gnomAD项目致力于提升其数据的多样性和代表性,从而能更全面地覆盖人类基因组的遗传变异。增加种群的代表性有助于更准确地理解和解释跨种族的遗传差异,特别是在疾病相关的遗传研究中。
集成长读测序数据
此外,TR-gnomAD项目的下一阶段计划优先整合那些通过长读测序(Long-Read Sequencing)新识别出的与疾病相关的串联重复序列(Tandem Repeats, TRs)。长读测序技术相比传统的短读测序(Short-Read Sequencing),能提供更长的读段,从而大幅提高对大片段串联重复序列的检测准确性和分辨率。通过整合这些数据,TR-gnomAD能够更有效地识别与疾病相关的TR扩张,进一步推动对遗传疾病的理解和诊断。这一策略是为了填补现有资源在识别疾病相关TR扩张方面的空白,为未来的医学研究和临床应用提供支持。
使用TR-gnomAD作为对照组的潜在问题
在使用TR-gnomAD数据库作为遗传疾病研究中的对照组时,为了确保研究结果的准确性和可靠性,需要格外注意病例-对照不匹配和取证偏见(ascertainment bias)。这些问题可能导致研究结论的偏差,进而影响疾病的遗传诊断和研究。
病例-对照不匹配(Case-Control Mismatch)
这一问题指的是在对照组和病例组的选择上存在的不一致性,如年龄、性别、种族和遗传背景等方面的差异。这些差异可能会影响到串联重复扩增(Tandem Repeat (TR) Expansions)的频率和表型,从而导致误解或错误的关联分析。
取证偏见(Ascertainment Bias):
取证偏见通常出现在样本选择过程中,特别是当研究的样本并非随机选择时。例如,如果某种疾病在特定人群中更常见,而该人群在数据库中的代表性不足,则可能导致对这种疾病相关TR的误解。
为了减少这些潜在问题的影响,研究人员应采取以下措施:
确保对照组和病例组的匹配:研究者应确保在遗传背景、年龄、性别等方面,病例组与对照组尽可能一致,以减少变异的非疾病相关因素的干扰。
增强样本的多样性和代表性:在构建和使用TR-gnomAD数据库时,应增加不同人种和地区的样本数量,尤其是那些在当前数据库中代表性不足的群体,以提高研究的普适性和准确性。
详细记录和分析潜在的偏见来源:在研究报告中详细记录所有可能的偏见来源和对研究结果的潜在影响,以便于其他研究者进行合理的解读和应用。
TR-gnomAD的潜在局限与未来研究方向
识别未知疾病风险的重复扩展的局限性
TR-gnomAD虽然提供了大规模的串联重复扩展(tandem repeat expansions, TRs)参考图谱,但目前还未能有效识别与未知疾病风险相关的新的TR扩展。尽管数据库覆盖了0.86百万个TRs,这些数据仅代表了人类基因组中总TRs的一部分,且多数来自已知的、可能与疾病无关的TRs。这表明尽管TR-gnomAD的数据基础坚实,但其在发现新的与疾病相关的TR扩展方面的能力还有待提高。
确定已知致病TRs的风险阈值
当前版本的TR-gnomAD在定义已知致病TRs(pathogenic TRs)的风险阈值方面存在局限。虽然通过比较疾病组与TR-gnomAD中的匹配祖先背景样本的TR单位数,TR-gnomAD可以作为控制队列用于解释已知的致病TRs,但是这种方法需要更多的临床数据来建立更精确的风险评估标准。这些风险阈值对于临床诊断和疾病预防至关重要。
未来的研究方向
为了解决这些问题,TR-gnomAD计划在未来的研究中分析更多的全基因组测序(whole-genome sequencing, WGS)数据,尤其是来自未充分代表的祖先背景的数据。此外,TR-gnomAD团队也计划优先鉴定那些通过长读测序(long-read sequencing)新识别为与疾病相关的TRs。通过结合受TRs影响的潜在患者队列进行更深入的分析,期望能够更全面地理解TRs与疾病的关系,从而提高TR-gnomAD数据库的临床应用价值
原文链接
Cui Y, Ye W, Li JS, Li JJ, Vilain E, Sallam T, Li W. A genome-wide spectrum of tandem repeat expansions in 338,963 humans. Cell. 2024 Mar 28:S0092-8674(24)00252-6. Epub ahead of print. PMID: 38582080.
DOI:https://doi.org/10.1016/j.cell.2024.03.004
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#全基因组测序# #TR-gnomAD# #串联重复#
11