
浙江大学王永成团队Angew:高通量宿主-微生物单细胞RNA测序技术
2024-03-05 BioMed科技 BioMed科技 发表于上海

scRandom-seq提供了一种高通量的跨物种双向单细胞RNA分析工具,将有助于未来发现并解开感染体系和肿瘤微环境等体系内的宿主-微生物相互作用的复杂关系。
近日,浙江大学良渚实验室王永成团队在国际学术期刊Angewandte Chemie在线发表“High-Throughput Host-Microbe Single-Cell RNA Sequencing Reveals Ferroptosis-Associated Heterogeneity during Acinetobacter baumannii Infection”的研究论文。
该研究首次建立了基于随机引物与微流控平台的高通量宿主-微生物双向单细胞转录组测序方案(scRandom-seq),并将其应用于鲍曼不动杆菌(A.b)感染THP-1诱导分化的巨噬细胞中,首次发现细胞内A.b驱动宿主细胞发生铁死亡,提示靶向铁死亡可以作为抵抗A.b感染的新策略。scRandom-seq提供了一种高通量的跨物种双向单细胞RNA分析工具,将有助于未来发现并解开感染体系和肿瘤微环境等体系内的宿主-微生物相互作用的复杂关系。

图1. 论文截屏
宿主与其内微生物组的相互作用对人类生理健康至关重要,宿主与病原体相遇的结果不仅取决于细菌的毒力特征,还取决于与宿主细胞的相互作用的时间点、位置和方式等。宿主-微生物相互作用的复杂性需要高分辨率的方法来捕捉它们从整个生物体到单个细胞各个尺度上的全部复杂性。最新的单细胞RNA测序(scRNA-seq)技术的突破开启了转录组学的新时代,发现了以前未知的细胞类型、生理状态和随机基因表达的原则。10X Genomics Chromium和BD Rhapsody平台已被广泛应用于真核生物,但它们都依赖于oligo(dT)-引物的逆转录(RT)反应对含poly-A尾 的RNA的捕获,不能捕获许多不含poly-A 尾的RNA类别,尤其是小的真核RNA。
最近,王永成团队开发了基于液滴微流控的FFPE样本单细胞全RNA测序技术和单细菌RNA测序技术,它们通过使用随机引物捕获真核和原核的全长总RNA,具有高通量、高灵敏度和高覆盖性等优点。此外,其他团队已开发了PETRI-seq、microSPLiT和BacDrop等用于细菌的scRNA-seq技术。由于宿主-微生物系统中宿主细胞丰富的RNA(约为微生物组的成百上千倍)且细胞与细菌之间的渗透差异,这些单细菌scRNA-seq技术无法直接用于研究复杂的宿主-微生物系统。尽管基于CEL-Seq2方法的scDual-Seq是一种可以捕获宿主和细菌转录组的单细胞双向RNA-seq方法,但它需要细胞分选和每个细胞的体外反应,具有操作复杂性和远未达到高通量水平的缺点。
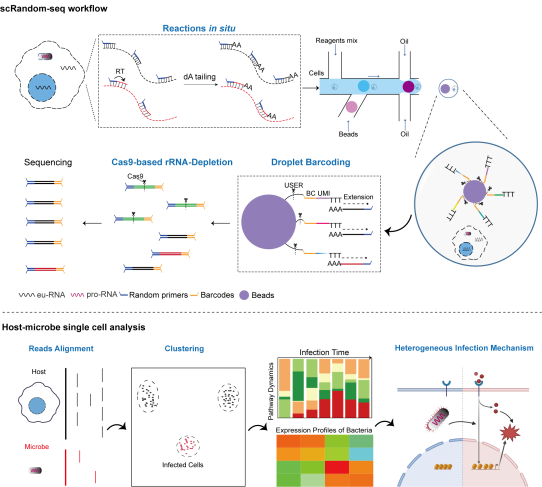
图2. 感染模型中scRandom-seq方法的示意图(选自论文,Figure 1)在本研究中,基于本团队前期新开发的基于随机引物的scRNA-seq化学体系,采用适当的透化处理、引入预索引策略、条形码设备优化、宿主rRNA去除和新的宿主-微生物单细胞分析管道等方面的改进,我们报告了一种基于半自动微流控的高通量的宿主-微生物双向scRNA-seq(scRandom-seq),以同时捕获真核和原核RNA,并试图在单细胞水平上研究宿主和病原微生物之间的相互作用和机制。(图2)
随后,我们使用参考细胞(人源的293T细胞和鼠源的3T3细胞的混合细胞)/细菌体系(A.b、大肠杆菌(E.coli)和肺炎克雷伯菌(K.p)的混合菌)对scRandom-seq的技术性能进行了评估,发现scRandom-seq均表现出高通量、高灵敏度和高覆盖性的优点,而基于CRISPR-Cas9的rRNA消解也展示出对人源rRNA高效的消解效果。(图3)
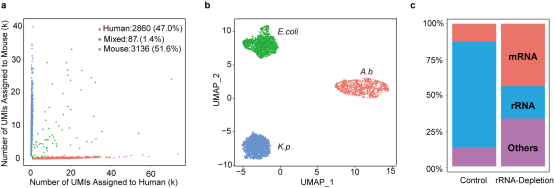
图3. scRandom-seq方法分别在混合的真核与原核样本中的性能(选自论文,Figure 2)
为进一步验证scRandom-seq在单细胞水平同时捕获宿主和病原体转录组的能力,我们使用了THP-1巨噬细胞(THP-Mø)模型,该模型中THP-1巨噬细胞以50:1的MOI(Multiplicity of Infection)被A.b感染0.5小时和4.0小时。通过scRandom-seq和基于Seurat的单细胞分析对THP-1来源的巨噬细胞和A.b的数据无偏比较分析,我们发现未感染、感染0.5小时和感染4.0小时的3个样品显示了四种表达模式(Cluster 1为未感染样品,Cluster 0为0.5小时感染样品,Cluster 2和Cluster 3(为4.0小时感染样品)。3个样品中检测到的A.b Reads显示了与CFU(Colony Forming Unit)测定从0小时到4.0小时的类似增长趋势。根据高度差异表达的基因(Differential Expressed Genes)以及KEGG富集分析,我们分别将Cluster 0的THP-1巨噬细胞称为“早期感染细胞”,Cluster 2和Cluster 3中的细胞称为“晚期感染细胞”。此外,使用Monocle 3对以上细胞进行拟时序的重编程轨迹分析,同样显示出细胞表达模式从未感染的细胞(Cluster 1)到“早期感染细胞”(Cluster 0)和“晚期感染细胞”(Cluster 2和Cluster 3)的感染模式的动态变化。(图4)
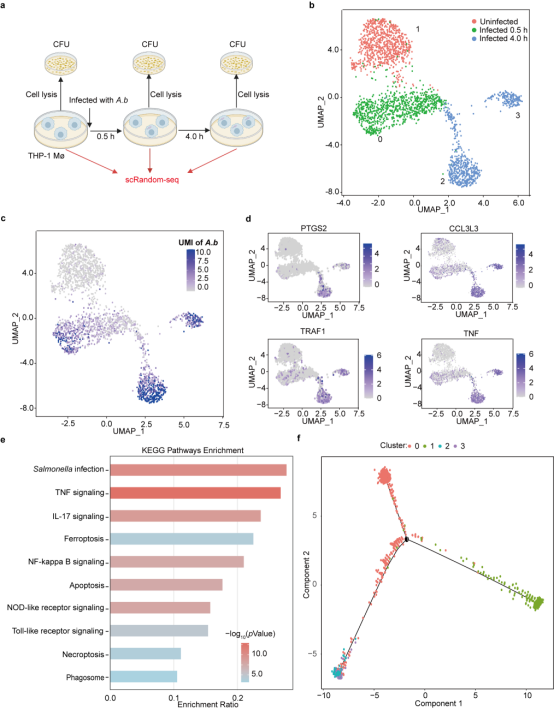
图4. scRandom-seq显示THP-Mø的极化与A.b感染有关(选自论文,Figure 3)
尽管Cluster 2和Cluster 3中的细胞都来自于4.0小时的样品,但它们确实具有不同的表达模式。因此,我们进一步使用它们的特定DEGs进行KEGG富集分析,以识别它们之间的差异。有趣的是,与Cluster 3相比,我们发现在Cluster 2中铁死亡通路和HIF-1信号通路更为突出。铁死亡是一种以铁过载和脂质过氧化为特征的新的程序性细胞死亡,也在假单胞菌、结核分枝杆菌、金黄色葡萄球菌和大肠杆菌感染中发现过。Cluster 2中与Cluster 3和Cluster 1(未感染样品)相比,铁死亡相关基因TFRC、SLC39A14、ACSL1、HMOX1(HO-1)、ACSL5和NCOA4在Cluster 2中更高表达,它们分别介导铁离子的胞内蓄积、铁蛋白自噬降解、脂质过氧化等,最终导致了宿主细胞的铁死亡。而Cluster 2和Cluster 3细胞来自同一个样本,因而我们假设Cluster 2细胞而不是Cluster 3细胞中的铁死亡压力可能归因于宿主细胞内A.b的不同表达模式。因此,我们整合每个Cluster 中的所有A.b Reads进行下游的相关分析。令人惊讶的是,Cluster 2和Cluster 3细胞中胞内A.b的表达模式完全不同。在Cluster 2细胞中,胞内A.b编码细菌铁蛋白(A1S-0800和A1S-3175)、铁硫蛋白(A1S-1512和A1S-3322)、铁载体受体蛋白(A1S-1725)和细胞分裂蛋白(A1S-2681和A1S-1930)的基因高度表达,这表明胞内A.b处于细胞分裂状态,对铁的需求更高。与Cluster 2中的宿主细胞不同,Cluster 2细胞中的胞内A.b中的谷胱甘肽过氧化物酶(gpx,铁死亡抑制剂)基因显著上调表达,这可抑制胞内细菌发生铁死亡。因此,我们提出以下图示,以展示胞内A.b对铁的需求如何驱动宿主细胞中的铁死亡压力以在体内生存甚至复制。(图5)
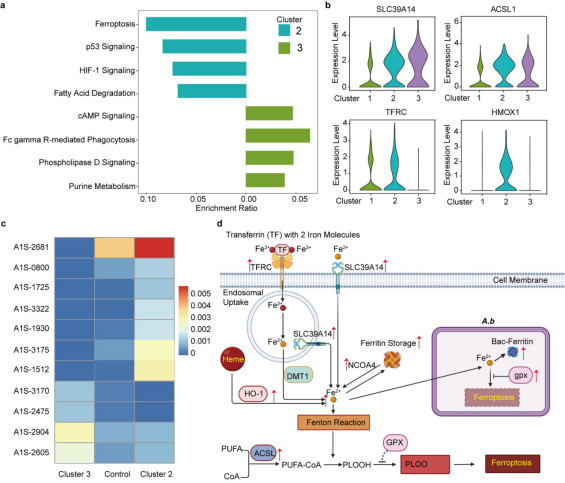
图5. 宿主微生物的铁死亡异质性是由胞内A.b引起的(选自论文,Figure4)
为验证上图中我们的假设,即铁死亡是否发生并使宿主细胞或胞内细菌受益,我们测试了铁死亡相关的标志物(铁离子蓄积相关的FTL和FTH1,氧化应激相关的谷胱甘肽与氧化谷胱甘肽(GSH/GSSG)比值和丙二醛(MDA)),以及后续对其进行铁死亡抑制剂(Ferrostatin-1,Fer-1)的处理。结果显示,感染4.0小时的THP- Mø样本,细胞活力更低、FTL蛋白显著增加、MDA含量显著增加、GSH/GSSG比值显著降低,宿主细胞确实存在铁死亡压力。而Fer-1处理可显著逆转这种压力,显著提高宿主细胞的细胞活力,最终增强了对细菌感染的抵抗力。因此,减轻宿主细胞的铁死亡压力可以有效抵抗A.b感染,可能成为治疗A.b感染的新靶点。(图6)
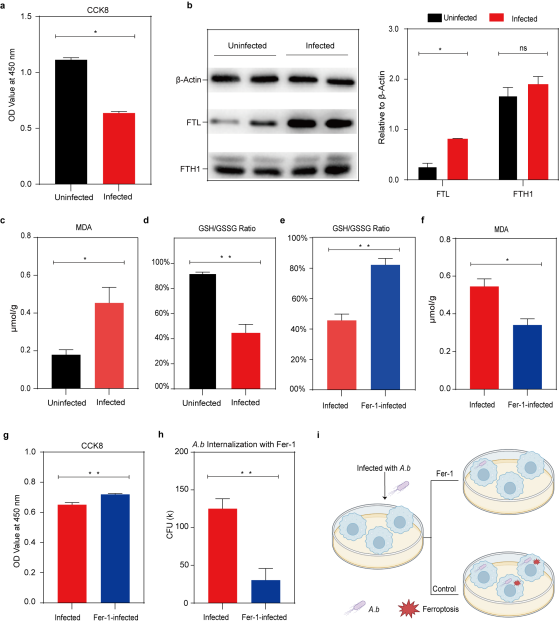
图6. Fer-1对铁死亡的抑制有助于宿主细胞抵抗A.b感染(选自论文,Figure 5)
综上所述,我们展示了scRandom-seq,一种新型的高通量宿主-微生物双向单细胞RNA测序技术,并评估了其在真核样本和不同细菌样本中的检测效能。将这项技术应用于A.b感染模型中,我们首次观察到宿主细胞中与铁死亡相关的异质性,这表明铁死亡可能成为抵抗A.b感染的新的临床靶点。scRandom-seq为研究感染模型和肿瘤-微生物样本中的宿主-微生物相互作用开辟了新的可能性,这有助于理解疾病的进展和制订新的精准的治疗策略。
浙江大学良渚实验室王永成研究员、浙江大学医学院附属邵逸夫医院华孝挺副研究员为本文的通讯作者。王永成团队孟红恩博士、张天宇,华孝挺团队张望博士为本文共同第一作者。浙江大学医学院附属邵逸夫医院的俞云松教授、陈航飞和浙江大学医学院王福俤教授、余盈盈博士,杭州跃真生物科技有限公司朱玉懿等也为本工作作出了贡献。研究获得了国家自然科学基金、浙江省领军型创新团队和浙江省尖兵研发攻关计划的支持。
参考文献
Meng H, Zhang T, Zhang W, Zhu Y, Yu Y, Chen J, Chen H, Wang F, Yu Y, Hua X, Wang Y. High-Throughput Host-Microbe Single-Cell RNA Sequencing Reveals Ferroptosis-Associated Heterogeneity during Acinetobacter baumannii Infection. Angew Chem Int Ed Engl. 2024 Feb 28:e202400538. doi: 10.1002/anie.202400538.
原文链接:
https://onlinelibrary.wiley.com/doi/10.1002/anie.202400538
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言













#RNA测序# #scRandom-seq#
20