先天性自愈性朗格汉斯组织细胞增生症一例
2017-10-22 中华皮肤科杂志 佚名

患儿男,1岁2个月。因头面部、躯干、四肢散在丘疹9个月,于2016年5月到广东省皮肤病医院门诊就诊。
【一般资料】
患儿男,1岁2个月
【主诉】
因头面部、躯干、四肢散在丘疹9个月,于2016年5月到广东省皮肤病医院门诊就诊。
【现病史】
患儿出生5个月时额部出现数个红色丘疹,可自行消退,但皮疹反复,此后躯干、四肢逐渐出现类似皮疹,散在分布,部分皮疹破溃、结痂,皮疹均可自行消退,消退后不留痕迹。
【体格检查】
各系统检查无异常。皮肤科检查:躯干四肢散在红色或皮色丘疹,突出皮面,表面光滑,边界清楚,触之质硬,无渗液,部分丘疹破溃结痂,黏膜部位未见皮疹(图1)。
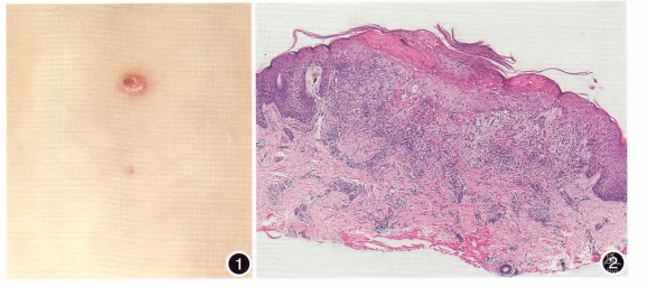
【辅助检查】
x线胸片、头颅正侧位片均未见异常,消化系统彩超未见异常。皮损组织病理:表皮灶性角化不全,见浆痂,基底细胞液化变性(图2),真皮浅层组织细胞,少许淋巴细胞,个别嗜酸性粒细胞浸润。免疫组化:CDla(+),S.100(+),Langrin(+)。
【初步诊断】
【治疗】
予0.03%他克莫司软膏局部外用,在随访中。
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#自愈#
40
#先天性#
36