
NBT:叶凯团队开发基于“序列-图像”转换的差异比较算法SVision-pro,实现高精确性SV检测及分型
2024-04-16 测序中国 测序中国 发表于上海

叶凯教授团队开发了SVision-pro,这是一种基于神经网络的实例分割框架,能够直观地表示基因组到基因组水平的测序差异,并在无需任何推断模型的条件下比较地发现基因组之间的SV。
基因组结构变异(SV)与生物性状进化和严重疾病表型密切相关,近年来,长读长测序技术(LRS)的快速发展促进了SV的检测,包括简单SV(SSV)和复杂SV(CSV)。特别地,新发(de novo)SV和体细胞SV是孟德尔疾病和癌症发生的主要原因,人们通常使用调用集合并策略(callset-merge)和read推断策略(例如nanomonsv)进行样本间基因组SV差异鉴定,但这些方法假阳性较高、应用局限性较大,检测时易出现、误报(尤其是CSV)。
2022年,西安交通大学叶凯教授团队开发了基于深度学习的多目标识别框架SVision,能够自动从长读长测序序列数据中检测和表征CSV。SVision可直接检测CSV,而不用匹配已知结构,灵敏地检测常见和以前未表征的复杂重排。虽然有了较大的进展,但目前基于深度学习的图像模式测序在SV比较发现的应用中仍受限,主要问题包括无法表征基因组间差异和需要同时检测基因组间SV和基因分型。
近日,叶凯教授团队在Nature Biotechnology发表了题为“De novo and somatic structural variant discovery with SVision-pro”的最新研究,展示了其在新生和体细胞SV鉴定方法方面取得的突破性进展。叶凯教授团队开发了SVision-pro,这是一种基于神经网络的实例分割框架,能够直观地表示基因组到基因组水平的测序差异,并在无需任何推断模型的条件下比较地发现基因组之间的SV。经评估,SVision-pro性能优于目前最先进的方法,与SV合并方法相比,其具有较低的孟德尔错误率,低频SV灵敏度高且假阳性率降低,可提高CSV的分辨率。

文章发表在Nature Biotechnology
多种遗传病和癌症的变异研究需要在多个样本之间进行基因组变异差异比较,相较与领域内常用的“先检测再求差”的分步式策略,叶凯教授团队提出了基于“序列-图像”转换策略的多样本差异比较算法SVision-pro,将SV的检测和分型问题从序列问题统一转化为图像空间的变异实例分割问题。
SVision-pro包括两个关键模块:序列-图像表示模块将两个样本的基因组特征编码在一张图像中,神经网络识别模块从中比较识别SV及其基因组间差异(图1)。SVision-pro集成了SV检测和基因组之间的基因分型,作为一个一站式的基于神经网络的图像实例分割任务,促进了新发及体细胞SSV、CSV的发现。
序列-图像表示模块首先将LRS数据中识别出的异常基因组位点作为输入,并将每个read结果总结为一系列符号,这些一维(1D)符号序列直接从read比对结果中获得,无需任何面向sv类型的预处理;随后,将上述符号迭代聚集在一起作为候选异常基因位点,该过程无需匹配已知的SV类型,确保了SV位点的全面捕获,特别是对于未开发的CSV。
接下来,序列-图像表示模块在结构草图(structure sketching)、内容渲染(content rendering)两个步骤中比较两个基因组(病例基因组及对照基因组)。结构草图步骤将1D read符号序列转换为2D相似性图像,内容渲染步骤用增强覆盖轨迹(ACT)填充稀疏图像区域,ACT表示病例和对照基因组之间的基因组差异。这种表示策略有助于基因组间的比较,同时编码SV结构及其基因组间差异。
神经网络识别模块将上述任务集成到一个基于多任务神经网络的图像实例分割框架中,对基因组中的SV进行分类和比较。该框架接收编码图像并生成像素级分割掩码,将图像区域分类为五个基本SV成分(component)类型、一个野生型参考(REF)和背景,SV类型是通过将病例和对照轨迹中的成分连接在一起直接预测的。此外,该框架还可以对病例和对照基因组之间的SV成分类型、断点和等位基因频率(AF)进行三任务比较。除了广泛使用的来自AF的基因分型标签,SVision-pro通过对比病例基因组与对照基因组中的SV成分,生成了四个不同的类型,即Germline、New component、New breakpoint和New alleles。综上,SVision-pro可为不同的灵敏度要求提供灵活的图像属性,目前最低检测AFs为0.01。
为确定合适的实例分割模型,研究团队在模拟数据上训练、比较了五个不同参数大小的模型,包括Unet、全卷积网络、Deeplabv3、Lite-Unet和mini-Unet(图1c)。最终,Lite-Unet在精度和模型大小之间实现了平衡,同时表现出很强的模型可解释性。
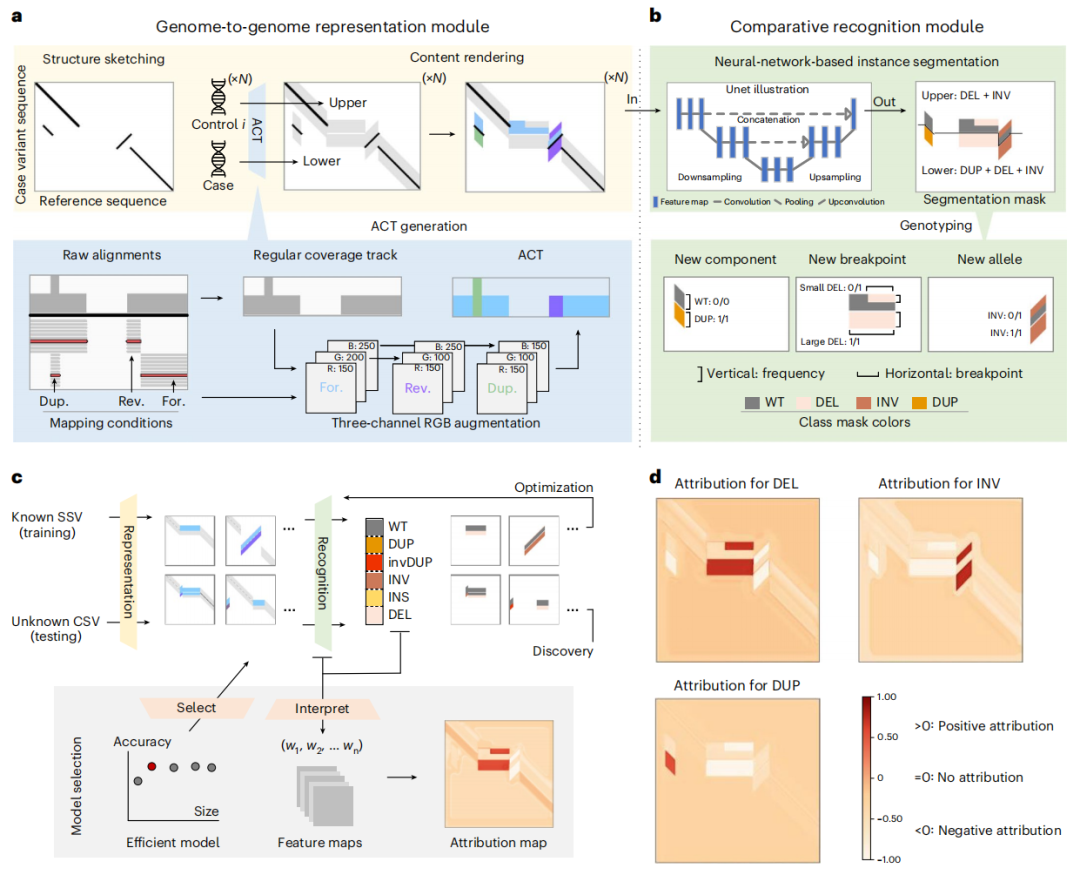
图1. SVision pro概述
研究团队使用模拟和公开可用的数据集对SVision-pro和其他方法的性能进行基准测试,包括HiFi、ONT和CLR。结果显示,SVision-pro在HG002真实SSV和模拟CSV上表现优于其他方法,在CSV子组件准确性方面达到96%-98%,与SVision相比平均提高了15%。进一步的实验证明,SVision-pro对CSV检测具有高灵敏度和低假阳性率。
在六个家系中,研究团队将SVision-pro与调用集合并策略(Sniffles2、SVision等)进行了比较(图2a)。结果显示,SVision-pro对六个家系进行了正确的基因分型,并在同卵双胞胎之间实现了最高的孟德尔一致性(HiFi read 99.3-98.4%,ONT read 94.5%-97.6%)和最低的不一致性(0.7%),优于其他方法。SVision-pro的高基因分型准确性还促进了孟德尔样本的可发现,特别地,其分型了包含两个SV等位基因的复杂基因位点:SSV(插入)和CSV(插入-删除)。
与其他模型相比,SVision-pro报告了26个新发SV,包括13个插入和13个缺失,所有这些SV都经过手动验证。上述结果表明,SVision-pro有效减少了孟德尔样本中的假阳性调用,并报告了高质量的新发SV。
研究团队使用正常-肿瘤配对细胞系HCC1395和HCC1395BL,通过三种测序技术(包括HiFi、ONT和CLR)评估了SVision-pro。SVision-pro检测到87%-90%已发表的体细胞SSV位点,Sniffles2检测到66-81%,nanomonsv检测到6%-29%(图2f)。使用Vapor28对检测到的体细胞调用进行验证发现,与Sniffles2(9.8%-40.3%)相比,SVision-pro的假阳性率更低(4.3%-8.7%)(图2g)。这些结果表明,SVision-pro在检测体细胞SV方面具有更高的灵敏度和更低的假阳性率。
最后,SVisionpro解析了先前被报告为SSV的8个CSV,并鉴定了一个非体细胞复杂基因位点,该基因位点先前被报道为体细胞SSV。
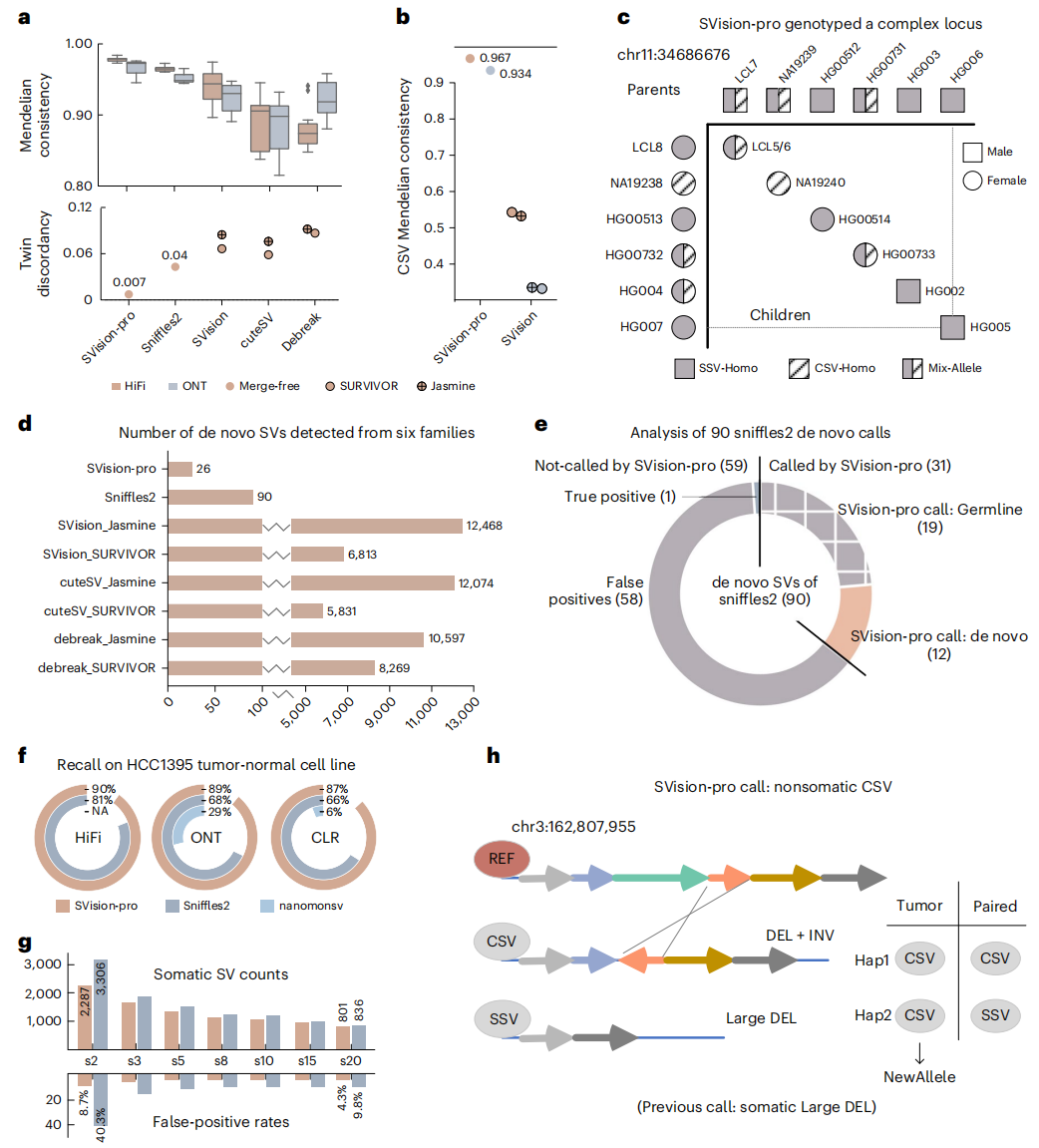
图2.性能比较
综上所述,SVisionpro是一种准确且可解释的方法,用于比较SV检测和基因分型;通过直观比较测序比对中编码的基因组特征,避免了调用集级策略容易出错的合并过程,产生高质量调用。SVisionpro实现了高精确性、低假阳性的新发SV和体细胞SV的精准识别,为后续从大规模专病队列数据、临床诊断数据中发现关键致病SV提供了关键技术支撑,并为基于“人工智能+” 的生物序列大数据计算框架提供了新思路。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#测序# #SVision-pro# #SV检测#
11