
Nat Commun:结合HiFi、Hi-C技术优势的新算法pstools,可高效、准确重建癌症染色体单倍型基因组
2023-05-06 测序中国 测序中国 发表于江苏省

该研究将高分辨率测序技术应用于COLO829癌症系,并开发了一种快速、准确的计算方法pstools。
癌症基因组是高度复杂且异质的,其携带多种复杂的结构变异(SV),标准的短读长测序和分析方法无法提供完整、精确的SV图谱。因此,为识别和表征突变,开发染色体水平(chromosome-scale)的单倍型基因组重建方法是十分重要的。迄今为止,已有大量研究(如ICGC-TCGA全基因组泛癌分析)使用短读长WGS数据报告了SV事件的发生,而高分辨率方法(特别是HiFi、Hi-C技术)在绘制综合构造变化图谱方面具有额外的优势,但基于高分辨率方法的研究设计尚不充足。
近日,丹麦技术大学Shilpa Garg团队在Nature Communications发表题为“Towards routine chromosome-scale haplotype-resolved reconstruction in cancer genomics”的文章。研究团队将高准确度的HiFi长读长测序和Hi-C长读长测序应用于黑色素瘤COLO829癌症细胞系中,并开发了一种高效的、基于图(Graph-Based)的pstools方法,可处理高分辨率的HiFi和Hi-C数据,用于染色体水平单倍型基因组重建,以精确表征癌症的SV情况。在没有亲本信息的情况下,pstools方法能在12h内生成染色体水平的高质量phased scaffold,优于现有方法。此外,该方法还在碱基分辨率下发现了准确的染色体水平SV。

文章发表于Nature Communications
研究团队通过精确的HiFi长读长、Hi-C长读长测序技术对黑色素瘤COLO829癌症细胞系进行测序(图1)。此外,研究团队还开发了一种基于图的计算工具pstools,可处理多种不同的数据类型,并将其整合在联合序列空间中,能够保留不同层次的基因组复杂性以直接产生完整的phased基因组。该方法还可以有效地表征重复区域中的SV,在碱基和单倍型分辨率上优化了癌性突变的特征,并且可以在全染色体水平上进行多倍体定相(phasing)。
研究团队将pstools方法应用于COLO829的体细胞和胚系细胞,在全基因组上以碱基分辨率发现了166个体细胞结构变异(SSV)。将其与使用其他技术观察到的SSV进行对比(图1b),结果显示,pstools方法在碱基和单倍型分辨率上提供了更精确的SV表征。上述结果表明,与短读长测序技术相比,pstools方法可以在重复区域中表征更精确、全面的单倍型SV图谱。
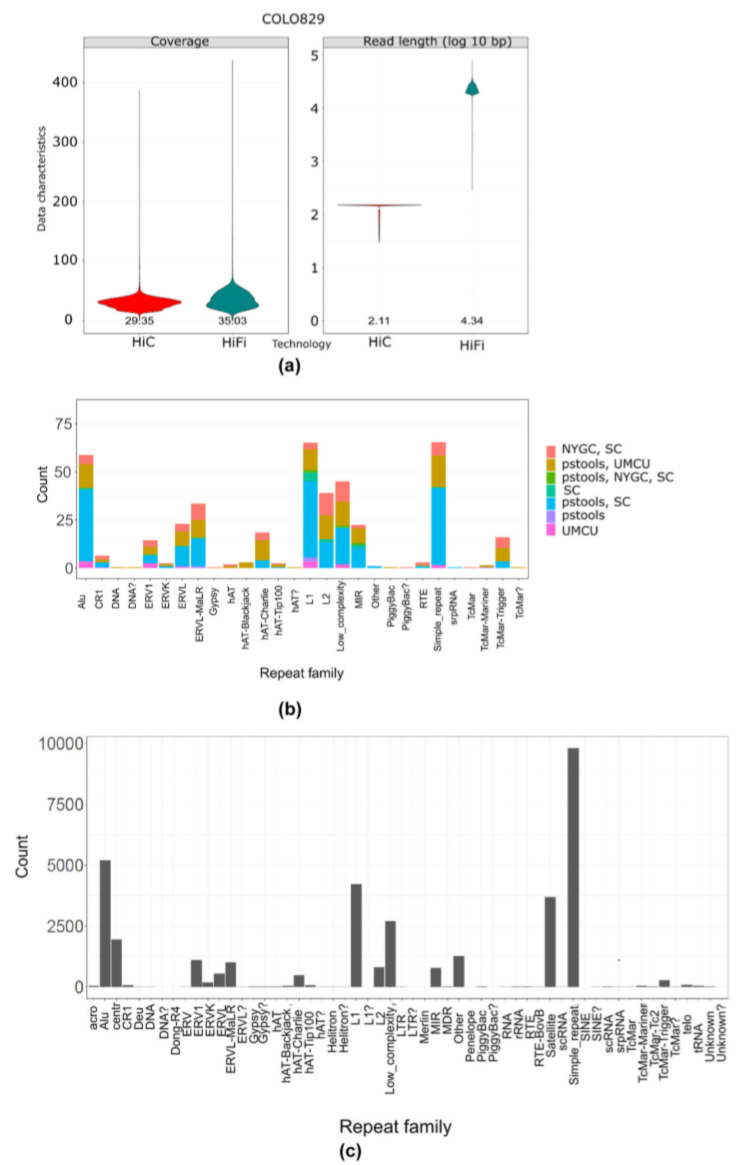
图1. COLO829 HiFi/Hi-C测序和SV的发现。来源:Nature Communications
图2详细介绍了pstools算法的工作流程,其有效结合了HiFi和Hi-C技术的优势,能准确地解离染色体以及连接染色体臂,可用于表征染色体间和染色体内结构序列事件,为常规临床应用生成准确、连续和完整的复杂癌症单倍型基因组。
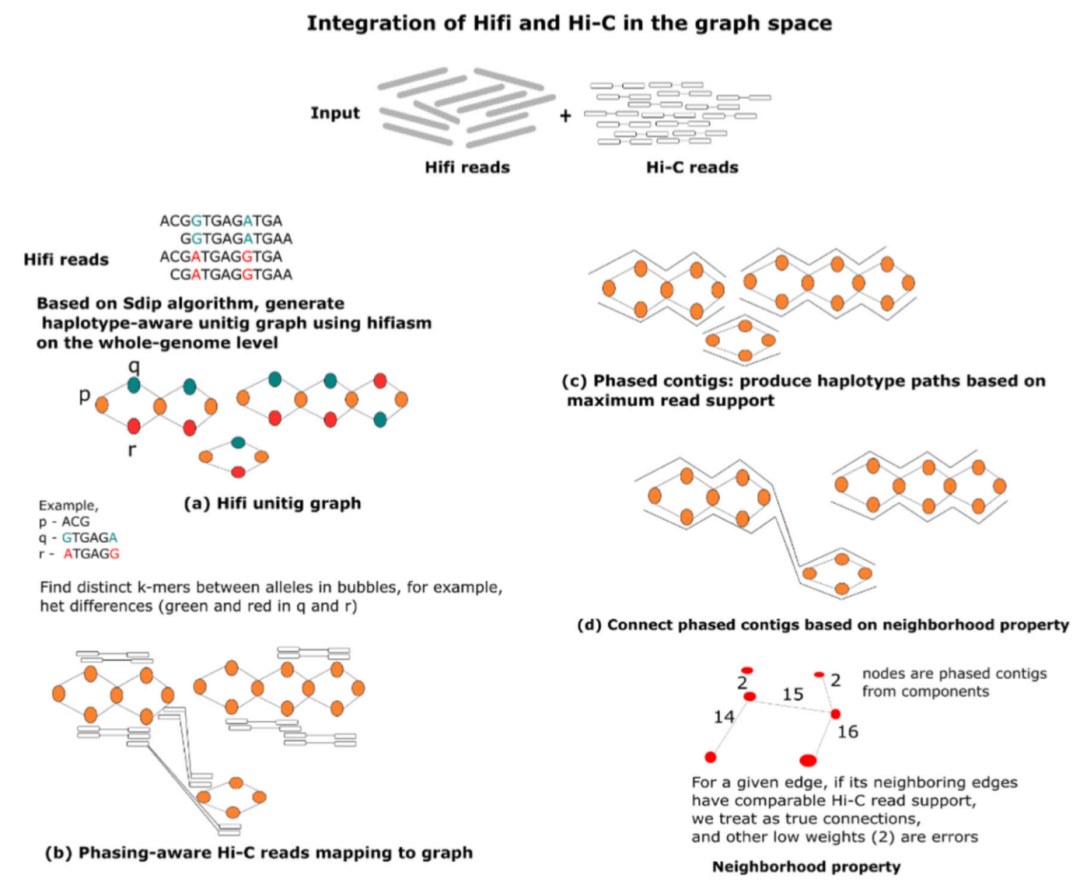
图2. Pstools算法的工作流程。来源:Nature Communications
研究团队在健康人体样本(HG002、HG00733和PGP1)上对pstools方法进行了基准测试,标准评估指标包括:NG50、switch/hamming误差以及总序列长度。结果显示,pstools算法生成了scaffold>6.0Gb的高质量组装 ,NG50组装>130 Mb。相比之下,使用hifiasm(Hi-C)方法生成的NG50组装<52 Mb,表明其不适用于染色体水平的基因组学研究。此外,pstools的组装质量较高,相位精度超98.5%,组装耗时较短,在12 h内就能完成(trio-hifiasm+salsa2方法耗时更长>2天)(表1)。
表1. phased序列评估统计表。来源:Nature Communications

为确认是否出现组装错误,研究团队使用Grch38作为参考序列,使用minimap2对上述组装进行比对操作,对scaffold进行了评估(图3)。结果显示,所有contig都被正确地分配到染色体中,与triohifiasm+salsa2相比,pstools方法可以产生高质量的phased scaffold。
研究团队还在COLO829癌症细胞系上对pstools方法进行了基准测试,并使用最先进的HiFi contigger(Hifiasm)和Hi-C scaffolde(salsa2)方法进行了独立对比实验。结果显示,Hifiasm+salsa2方法不能在染色体水平上重建phased基因组。相比之下,pstools方法不需要任何亲本信息,就能在染色体水平上产生兼具完整性、准确性和连续性的高质量组装(NG50 > 130 Mb)。此外,在碱基分辨率下,研究团队通过pstools共发现了19,956个插入、14,846个缺失、421个重复、52个倒置和498个易位,表明其能够鉴定复杂癌症基因组的单倍型SV(胚系和体细胞)并进行表征。

图3. HG002(左)和COLO829(右)的phased序列图。来源:Nature Communications
研究团队将通过pstools方法得到的高置信度SV调用集与其他多种技术(PacBio-CLR、Nanopore、PacBio-HiFi和短读长测序)得到的SV调用集进行了比较。结果显示,pstools方法的F1评分为93.9%(精确率96.0%,召回率91.9%),而基于Dipasm、Hifiasm+salsa2和Hifiasm+3D-DNA方法的F1评分<82%。与现有方法相比,pstools方法在癌症基因组学研究中更具优势。
有趣的是,pstools方法还能够检测到10号染色体上PTEN的纯合12kbp缺失(图4),可以发现由于同一或不同染色体上多个事件组合而产生的“断裂-融合-桥”(BFB)循环事件的SV。例如,研究团队在第15号染色体上发现了一个已知的BFB事件,该事件来自第6号和第20号染色体的插入。

图4. COLO829的全基因组SV表征。来源:Nature Communications
综上所述,该研究将高分辨率测序技术(HiFi和Hi-C)应用于COLO829癌症系,并开发了一种快速、准确的计算方法pstools,其性能优于现有方法trio-hifiasm和salsa2,有助于精确发现SV、重建染色体水平单倍型基因组,为分析单个患者的全谱SV提供了有效且简化的方法。此外,pstools方法能够产生高质量的胚系SV调用集,为临床医生提供了一种简单的方法来分析患者的SV,有助于更好地对患者进行诊断和疾病管理,包括预测治疗反应。
参考文献:
Garg S. Towards routine chromosome-scale haplotype-resolved reconstruction in cancer genomics. Nat Commun. 2023;14(1):1358. Published 2023 Mar 13. doi:10.1038/s41467-023-36689-5
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




谢谢分享
39