如何搞懂淋巴浆细胞性淋巴瘤?这篇文章是我看到最棒的梳理!
2022-11-23 医学术 医学术

淋巴浆细胞性淋巴瘤(lymphoplasmacytic lymphoma, LPL,旧称淋巴浆细胞样淋巴瘤)是一种罕见的成熟B细胞淋巴瘤,常侵犯骨髓,少数情况下也可侵犯脾脏和/或淋巴结。
淋巴浆细胞性淋巴瘤(lymphoplasmacytic lymphoma, LPL,旧称淋巴浆细胞样淋巴瘤)是一种罕见的成熟B细胞淋巴瘤,常侵犯骨髓,少数情况下也可侵犯脾脏和/或淋巴结。华氏巨球蛋白血症(Waldenström macroglobulinemia, WM)是一种具有临床病理性特征的疾病,与血液中IgM型单克隆丙种球蛋白病相关,LPL患者几乎都有该表现。

本文将总结LPL的流行病学、病理生物学、临床表现、病理特征、诊断及鉴别诊断。
在美国及西欧,LPL约占血液系统恶性肿瘤的1%,其发病率大约为每年8.3/1,000,000,亚洲的发病率约为美国和西欧的十分之一。绝大多数患者为白种人,其他族群约占所有病例的5%。患者获得诊断时的中位年龄为65岁,50%-60%的患者为男性。
大部分LPL患者的循环中存在单克隆IgM,常可导致一种高黏滞综合征,称为WM。虽然大多数情况下WM似乎表现为散发性疾病,但某些病例呈现出家族易感性LPL和WM的流行病学,此处不做更详细的讨论。
偶有归类为LPL的病例与混合型(Ⅱ型)冷球蛋白血症伴丙肝病毒(hepatitis C virus, HCV)感染有关。然而,很多此类病例实际上可能属于脾边缘区淋巴瘤(marginal zone lymphoma, MZL)的诊断范畴,因为MZL也与HCV感染相关,且可能伴有异常蛋白血症。
LPL中的恶性细胞推测来源于外周B淋巴细胞(受到刺激后可分化为浆细胞),可能是对抗原初次免疫应答后的未进入生发中心的B细胞,或者是在生发中心经历了体细胞突变而未发生抗体类别(重链)转换的B细胞。一项基因表达谱分析研究将WM的转录谱与多发性骨髓瘤及慢性淋巴细胞白血病(chronic lymphocytic leukemia, CLL)的转录谱进行了对比,结果显示WM的表型更类似于CLL。
LPL的发病机制尚未完全理解,但和其他淋巴瘤一样,获得性遗传改变和表观遗传改变似乎都起作用。
一项研究纳入14例LPL患者,发现其中有13例的免疫球蛋白重链(immunoglobulin heavy chain, IgH)基因发生了体细胞高频突变,而未发生克隆内变异(intraclonal variation),提示LPL细胞起源于在生发中心经历了亲和力成熟的B细胞。其他几项研究发现,LPL中IgH位点的染色体重排并不常见。然而,40%-60%的WM患者中识别出了6q21-q25缺失,6q21-q25也是多种B细胞淋巴瘤的一个常见染色体丢失位点。6q21-q25区的致病性靶基因尚未确定,但很显然,在经常缺失的区域中,包括了编码NF-κ-B、BCL2、细胞凋亡和浆细胞分化调节因子的基因。
新一代LPL基因测序技术在以下基因中识别出了常见的频发突变:MYD88(95%-97%)、CXCR4(30%-40%)、ARID1A(17%)和CD79B(8%-15%)。CXCR4基因的突变与WHIM综合征中类似,WHIM即:疣(warts)、低丙种球蛋白血症(hypogammaglobulinemia)、感染(infection)及先天性骨髓粒细胞缺乏症(myelokathexis)。多项研究显示,体细胞突变状态可将肿瘤分为临床表现和生存率各不相同的组群。与MYD88突变型肿瘤相比,MYD88野生型肿瘤的病程更具侵袭性,患者总生存期更短,依鲁替尼治疗效果较差。同时具有MYD88和CXCR4突变的肿瘤更可能表现为高黏滞血症和骨髓受累,并且相对于CXCR4野生型肿瘤,其获得重大疗效所需的时间更长,疗效达到的深度更浅,无进展生存率更低。
另一项报告整合了突变数据与全基因组DNA甲基化和转录组分析结果,识别出了2个具有不同DNA甲基化模式的LPL亚类,一个类似于记忆性B细胞,另一个类似于浆细胞。记忆细胞样肿瘤与CXCR4突变、13q缺失、脾肿大和血小板减少有关,而浆细胞样肿瘤与6q缺失、使用IGHV3可变区基因、CD38和其他浆细胞标志物的表达有关。
MYD88突变
MYD88是一个在Toll样受体及IL-1受体信号传递中发挥一定作用的分子,可增强B细胞的存活。LPL的发病机制涉及MYD88的一个激活性点突变(MYD88 L265P)。存在MYD88 L265P突变的LPL细胞中,MYD88与Bruton酪氨酸激酶(Bruton tyrosine kinase, BTK)的复合物可促进肿瘤的生存。
一项研究对同一患者的骨髓LPL细胞及与之匹配的正常细胞进行全基因组大规模平行测序,发现10例LPL患者的骨髓LPL细胞中均存在MYD88 L265P突变。在其他患者中对该基因进行测序发现,54例WM患者中有49例存在MYD88 L265P突变,3例非IgM分泌型LPL患者则全部存在MYD88 L265P突变。而LPL患者的的正常组织样本以及10例健康人和10例多发性骨髓瘤患者的B细胞中均不存在该突变。MYD88 L265P突变在MZL和IgM型意义未明的单克隆丙种球蛋白血症(monoclonal gammopathy of undetermined significance, MGUS)中也非常少见,仅有7%的MZL患者和10%的MGUS患者有该突变。
随后的一项研究发现27例WM/LPL患者中有18例(67%)、28例结外MZL患者中有2例(7%)、53例脾MZL患者中有2例(4%)存在MYD88 L265P突变,而在11例结内MZL患者中无一例存在MYD88 L265P突变。
第3项研究发现,104例WM患者中有97例(93%)、24例IgM型MGUS患者中有13例(54%)存在MYD88 L265P突变。相比之下,20例脾MZL患者中只有2例、26例慢性淋巴细胞性淋巴瘤患者中只有1例存在该突变,而在多发性骨髓瘤患者、IgG型MGUS患者及健康人中均不存在该突变。
另一项研究纳入了64例伴巨球蛋白血症的MYD88野生型B细胞肿瘤患者,系统性回顾了其临床病理特征。结果显示,高达30%的病例有LPL以外的诊断,包括IgM多发性骨髓瘤和弥漫性大B细胞淋巴瘤。MYD88野生型WM病例的10年生存率低于MYD88 L265P突变型病例(73% vs 90%)。
总之,这些发现提示MYD88突变在LPL的发病机制中发挥核心作用。MYD88可增强Toll样受体的信号传递,从而激活NF-kB家族的转录因子,这些因子与正常B细胞或肿瘤性B细胞的生长及存活有关。
如上文所述,MYD88似乎还可通过BTK途径增强信号传递,BTK是B细胞受体信号通路中的重要组成部分,可促进B细胞的存活及生长。MYD88突变虽然与LPL高度相关,但并不是完全特异性的;据报道,在弥漫性大B细胞淋巴瘤的一个亚型中也存在这种突变,并且如上所述,在其他浆细胞病的一个亚型(如IgM型MGUS)以及低级别B淋巴细胞增生性疾病中也存在这种突变。
临床特征LPL患者的临床表现各不相同,可表现为与肿瘤浸润有关的症状(淋巴结肿大、脏器肿大及血细胞减少)或与单克隆蛋白生成有关的症状(高黏滞血症和神经病变)。大约1/3的患者在诊断时无临床症状。最常见的起病特征包括无力和疲乏,常是由贫血引起。多达1/4的患者可出现全身性B症状(即发热、盗汗及体重减轻)。
淋巴结肿大、肝肿大和脾肿大分别见于约20%的患者。70%以上的患者在诊断时即为淋巴瘤Ⅳ期,因为骨髓已受累。

丙种球蛋白病
大多数患者的血清在进行蛋白电泳或免疫固定电泳后可检出单克隆Ig(丙种球蛋白病),但存在丙种球蛋白病并不是诊断LPL所必需的。血清单克隆IgM是最常见的亚型,据此可诊断WM临床综合征。少见情况下,肿瘤可产生其他Ig、Ig组合(即,IgM和IgG)、混合性冷球蛋白或γ重链。如前所述,多数混合性冷球蛋白血症病例可与并存的HCV感染相关。
小部分患者的肿瘤细胞可分泌IgG和/或在细胞膜表面表达IgG,除此之外符合典型的LPL。一项病例系列研究显示,这类患者的中位年龄为70岁、存在明显的淋巴细胞增多和脾肿大、CD79b呈强阳性、12号染色体三体的发生率为60%,临床病程为非侵袭性。
与其他副蛋白血症病例一样,血清总蛋白水平和红细胞沉降率通常升高,并且实验室检测还可能出现伪差。
其他
诊断时存在轻度贫血的患者可达40%。严重贫血、中性粒细胞减少及血小板减少的情况非常少见。β2-微球蛋白可升高。血清乳酸脱氢酶通常正常。
循环中的单克隆蛋白可能干扰实验室检测,从而导致伪差。这部分内容不做深入展开讨论。
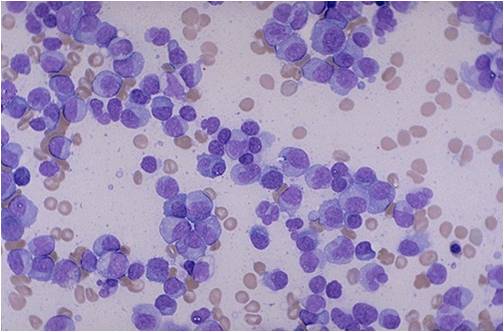
LPL的浸润细胞通常包含小B细胞、浆细胞样淋巴细胞及浆细胞,可累及骨髓,少数情况下可累及淋巴结和/或脾脏。在外周血中,罕见循环肿瘤细胞。
组织学
1、骨髓
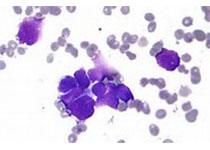
骨髓浸润可能为弥漫性、结节性或间质性,伴或不伴骨小梁旁聚集。浸润细胞包含小淋巴细胞、浆细胞和浆细胞样细胞,细胞数量不定,常混有反应性肥大细胞,也可能混有数量不定的免疫母细胞。

部分细胞的胞质Ig(IgM)积聚形成PAS染色(Periodic Acid-Schiff stain)阳性的包涵体,根据其在细胞内的定位,称为Russell小体(在细胞质内)或Dutcher小体(在伪核内)。
细胞外Ig沉积可表现为无定形体或晶体,有时伴有异物巨细胞反应。此类沉积物通常为偶然发现,没有临床意义。LPL患者可能发生淀粉样沉积,但不如骨髓瘤患者和其他浆细胞肿瘤患者常见。
2、淋巴结
淋巴结结构常得以保留,但可因小淋巴细胞、浆细胞和浆细胞样细胞的弥漫性、间质性浸润而消失。某些病例也可能含有较大的免疫母细胞样细胞。淋巴窦通常是开放的,窦腔内可能含有一些特殊的组织细胞,可与分泌的PAS染色阳性的Ig起反应。典型特征包括Dutcher小体和Russell小体、肥大细胞及含铁血黄素细胞。罕见情况下,可见大量病因未知的非干酪性肉芽肿。在某些病例中,此类非干酪性肉芽肿可能非常多,以至于潜在的淋巴瘤不明显甚至被忽略。
重要的是,淋巴结中不存在增殖中心[慢性淋巴细胞白血病/小淋巴细胞白血病(chronic lymphocytic leukemia/small lymphocytic leukemia, CLL/SLL)标志],也不存在灰白样边缘区分化(见于MZL)。
3、脾脏
脾脏的红髓及白髓均可被浸润。其浸润模式通常为弥散性,红髓中没有明显的边缘区或结节状。
4、外周血
循环中的恶性肿瘤细胞通常呈浆细胞样外观(即类似于浆细胞)。此类细胞通常为椭圆形,且含有丰富的嗜碱性细胞质。细胞核为圆形,其位置偏心,并存在核周凹陷或透明细胞质。细胞核含有“钟面”或“车轮辐条状”染色质,但没有核仁。
免疫表型
LPL细胞表达高水平的表面免疫球蛋白(surface immunoglobulin, sIg),通常为IgM型且通常缺乏表面IgD。目前已报道了表达IgG和IgA的病例。
这些肿瘤中浸润的淋巴细胞会表达泛B细胞抗原(CD19、CD20、CD22、CD79a),而浸润的浆细胞通常会下调CD20并上调CD138的表达。大多数可表达淋巴细胞功能相关抗原1(lymphocyte function associated antigen 1, LFA-1)。约一半肿瘤可表达粘附分子L-选择素、ICAM-1、CD44和CD11c。这些细胞的CD43表达情况不一;在某些病例中,CD25或CD11c可能呈弱阳性。少数病例可表达CD5。通常不表达CD10、CD103及CD23。
遗传学特征
如前所述,最常见的遗传学畸变为MYD88点突变。LPL无特异性染色体异常。肿瘤细胞的Ig重链及轻链基因发生了重排,可变区(variable region, V区)基因发生了体细胞突变,这提示这些细胞来自于一个经历过抗原驱动性选择的B细胞群。
染色体6q21-q25缺失是最常见的染色体拷贝数异常,可见于一半以上累及骨髓的病例,但在累及淋巴结的病例中较少见。不太常见的染色体改变包括:3号染色体三体、8号染色体三体及4号染色体三体。染色体易位t(9;14)(p13;q32)最初有一些病例报道,但随后发现这种易位在LPL中罕见且不具有特异性。
诊断
诊断的依据为受累组织的病理评估,通常为骨髓或淋巴结。联合组织学和免疫表型检查结果可排除其他伴浆细胞分化的小B细胞淋巴组织肿瘤:
必须有≥10%的活检样本显示小淋巴细胞、浆细胞样淋巴细胞和浆细胞浸润,且混合有数量不定的免疫母细胞。骨髓中的肥大细胞会出现特征性(但不具有诊断意义)增生,并伴有肿瘤性浸润。不存在增殖中心(CLL/SLL的诊断特征)及灰白样边缘区分化(见于MZL)。
这种浸润应表达一种典型的免疫表型(如,表面IgM+、CD5-/+、CD10-、CD19+、CD20+、CD22+、CD23-、CD25+、CD27+、FMC7+、CD103-、CD138-)。浆细胞性成分应该是CD138+、CD38+和CD45-/dim。
可能存在典型免疫表型的变化,但这种考虑的目的是合理排除其他淋巴细胞增生性疾病。检测MYD88 L265P突变有助于疑难病例的诊断。MYD88突变符合LPL,但不具有特异性。相反,无MYD88突变的病例不太可能是LPL。
LPL患者的进一步评估包括血清蛋白电泳(serum protein electrophoresis, SPEP),以确定是否存在单克隆Ig“峰”,该现象与WM临床病理有关。
鉴别诊断LPL的鉴别诊断包括伴浆细胞分化的其他小B细胞淋巴组织肿瘤。
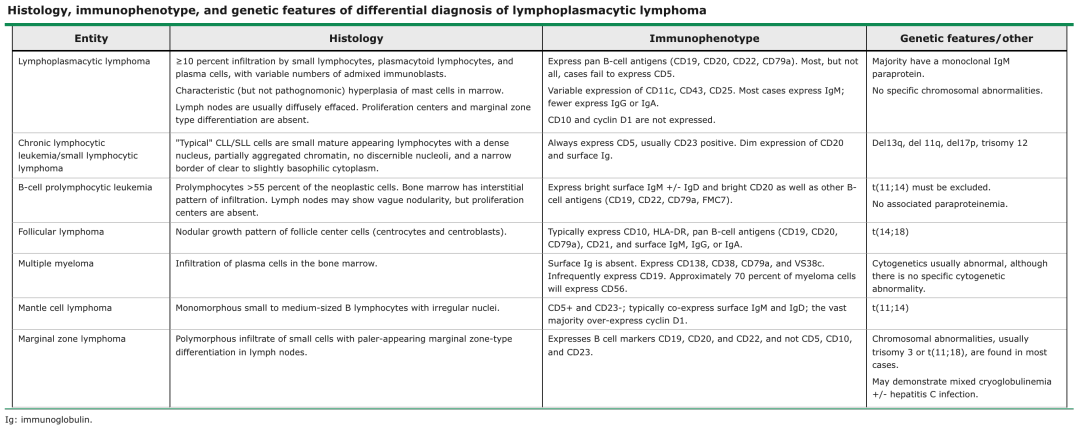
1、慢性淋巴细胞白血病
LPL和CLL/SLL都是小淋巴细胞增生性疾病,其病程进展缓慢。有助于区别CLL与LPL的特征包括:
外周血受累一般在CLL中更显著,但极少数情况下LPL也可出现“白血病样”表现。LPL患者循环肿瘤细胞通常呈浆细胞样外观,而CLL患者循环肿瘤细胞通常类似成熟淋巴细胞混合有数量不定的幼淋巴细胞(有单个明显核仁的较大的细胞)。
LPL患者的骨髓浸润通常不如在CLL中广泛,且与CLL不同的是,LPL患者的浸润细胞包括小淋巴细胞、浆细胞及浆细胞样细胞,但缺乏幼淋巴细胞和增殖中心。频繁出现反应性肥大细胞也是LPL的特征,但有时也可见于CLL。
LPL中受累的淋巴结缺乏CLL/SLL的诊断特征—增殖中心。
LPL具有下列免疫表型特点:CD23阴性、表面IgM和CD20强阳性、胞质Ig阳性。大多数LPL病例缺乏CD5的表达,而CD5可在CLL中表达。
2、多发性骨髓瘤
多发性骨髓瘤是一种浆细胞肿瘤,其特征为骨髓中存在恶性浆细胞,血清或尿液中存在单克隆蛋白。与高黏滞血症有关的症状很少见。不同于LPL,存在IgM副蛋白的典型多发性骨髓瘤极为少见,在所有患者中占比不足1%。
一般而言,LPL缺乏CD56、有大量小淋巴细胞表达克隆性表面Ig,因此可与多发性骨髓瘤相区分。在疑难病例中,可能必须根据临床表现的差异来排除多发性骨髓瘤。例如,如果存在溶骨性病变(伴或不伴高钙血症),应优先考虑IgM型多发性骨髓瘤,而不是LPL。如果存在高黏滞血症的症状,并且存在淋巴结肿大和/或脾肿大,则优先考虑LPL。
3、套细胞淋巴瘤
套细胞淋巴瘤(Mantle cell lymphoma, MCL)通常由小到中等大小的单形B淋巴细胞组成,细胞核形状不规则。MCL肿瘤细胞通常为CD5+及CD23-;绝大多数会过表达细胞周期蛋白D1,可通过免疫组化检测确定。通过传统的细胞遗传学检查,在略超过半数的MCL患者中可检出涉及细胞周期蛋白D1基因(cyclin D1 gene, CCDN1)的染色体易位t(11;14),但采用FISH技术检出的患者比例要高得多。该易位可见于某些多发性骨髓瘤患者,但不会见于LPL患者。
4、边缘区淋巴瘤
LPL和MZL都是小细胞多形性浸润的肿瘤。其免疫表型也与MZL细胞相似,即表达B细胞标志物CD19、CD20及CD22,但不表达CD5、CD10及CD23。大多数病例都存在染色体异常,通常为3号染色体三体或t(11;18)。
不同于LPL,MZL通常有一个富含淡染细胞质的细胞群(所谓的单核细胞样B细胞),这提示存在边缘区分化。此外,MZL患者更可能被证实存在混合冷球蛋白血症及HCV感染。然而,由于LPL与MZL的组织学及免疫表型相似,可能仍难以对二者进行鉴别。虽然在MZL中可能存在IgM副蛋白,但IgM的水平通常低于0.5g/dL,而大多数LPL患者的IgM副蛋白水平可超过0.5g/dL。某些中心正在开展对MYD88突变的检测,这也可能有用,但如前所述,这并不是一个绝对的鉴别点,因为小部分MZL也存在MYD88突变。对于某些疑难病例,先归类为“小B细胞淋巴瘤伴浆细胞性分化”,之后再纳入LPL和MZL进行鉴别诊断,这可能是最准确的分类方法。
5、滤泡淋巴瘤
罕见情况下,滤泡性淋巴瘤(follicular lymphoma, FL)可表现出弥漫性生长模式,并可出现一定程度的浆细胞样分化。与FL不同,LPL呈CD10阴性,也没有大多数FL病例中涉及BCL-2的染色体易位。
预后LPL的临床病程进展缓慢,欧洲的一些病例系列研究报道,LPL比典型的CLL/SLL更具侵袭性,患者的中位生存期为5-7年。然而,在REAL临床研究中,LPL患者的5年总体生存率(58%)和无失败生存率(25%)与CLL/SLL患者相同。最初的研究显示,无MYD88突变与淋巴结肿大及预后较差相关,但这种相关性需要进一步的研究。
大多数LPL患者存在循环单克隆IgM,符合WM的诊断。
淋巴浆细胞性淋巴瘤(LPL)是一种不常见的成熟B细胞淋巴瘤,通常累及骨髓,较少数情况下可累及脾脏和/或淋巴结。华氏巨球蛋白血症(WM)是一种具有独特临床病理特征的疾病,其骨髓表现为LPL且血液表现为IgM型单克隆丙种球蛋白病。
LPL的临床表现各不相同,包括与肿瘤浸润有关的症状(淋巴结肿大、脏器肿大及血细胞减少)或与单克隆蛋白生成有关的症状(高黏滞血症、神经病变)。大约1/3的患者无症状。
大多数患者都证实存在单克隆丙种球蛋白病,但丙种球蛋白病并不是诊断LPL所必需的。最常见的亚型为单克隆IgM ,据此可诊断WM。少见情况下,肿瘤可产生其他Ig、Ig组合、混合性冷球蛋白或γ重链。
LPL的诊断依据为受累组织的病理评估,通常为骨髓或淋巴结。联合组织学和免疫表型检查结果,可排除其他伴浆细胞性分化的小B细胞淋巴组织肿瘤:
1.必须有≥10%的活检样本证实存在小淋巴细胞、浆细胞样淋巴细胞和浆细胞的浸润,且混合有数量不定的免疫母细胞。在骨髓中,肥大细胞存在特征性(但不具有诊断意义)增生,并伴肿瘤性浸润。增殖中心[慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(CLL/SLL)的诊断特征]以及显示边缘区分化的富含淡染细胞质的细胞[所谓的单核细胞样B细胞,见于边缘区淋巴瘤(MZL)]在LPL中都不存在。
2.这种浸润应表达一种典型的免疫表型(如,表面IgM+、CD5-/+、CD10-、CD19+、CD20+、CD22+、CD23-、CD25+、CD27+、FMC7+、CD103-、CD138-)。浆细胞性成分应该是CD138+、CD38+和CD45-/dim。
可能存在典型组织学和免疫表型的变化,但这种考虑的目的是合理排除其他淋巴细胞增生性疾病。
检测MYD88 L265P突变对于疑难病例有诊断价值。
淋巴浆细胞性淋巴瘤(LPL)患者的进一步评估包括血清蛋白电泳(SPEP),以评估是否存在与WM有关的单克隆Ig“峰”。
LPL的临床病程进展缓慢,类似于典型的慢性淋巴细胞白血病/小淋巴细胞白血病(CLL/SLL)。
参考文献:
Arnold S Freedman, MD,Jon C Aster, MD, PhD,Clinical manifestations, pathologic features, and diagnosis of lymphoplasmacytic lymphoma
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言