综述:ACE2的主要功能以及在新型冠状病毒中作用
2020-02-02 CK医学科普 CK医学科普

血管紧张素转换酶(ACE) 2是羧肽酶ACE的同源物,羧肽酶生成血管紧张素II,这是肾素-血管紧张素系统(RAS)的主要活性肽。在2000年克隆ACE2之后,迄今为止已经描述了三种主要的ACE2功能。首先,ACE2已经成为RAS的一个强力负调节因子,可平衡ACE的多种功能。通过靶向血管紧张素II,ACE2在心血管系统和许多其他器官中显示出保护作用。第二种ACE2被鉴定为引起SARS冠状病毒也是此次
血管紧张素转换酶(ACE) 2是羧肽酶ACE的同源物,羧肽酶生成血管紧张素II,这是肾素-血管紧张素系统(RAS)的主要活性肽。在2000年克隆ACE2之后,迄今为止已经描述了三种主要的ACE2功能。
-
首先,ACE2已经成为RAS的一个强力负调节因子,可平衡ACE的多种功能。通过靶向血管紧张素II,ACE2在心血管系统和许多其他器官中显示出保护作用。
-
第二种ACE2被鉴定为引起SARS冠状病毒也是此次2019新冠状病毒的基本受体,而在SARS中,ACE2的下调在病毒感染后严重肺衰竭的发病机制中起着重要作用,关于新冠状病毒经由ACE2的研究文献见前述链接。
-
第三,ACE2及其同源物Collectrin均可与氨基酸转运蛋白结合,并在肾脏和肠道对氨基酸的吸收中发挥重要作用。
1.介绍
肾素-血管紧张素系统(RAS)在维持血压稳态以及哺乳动物体内体液和盐平衡方面起着关键作用。RAS的异常激活与心血管和肾脏疾病如高血压、心肌梗死和心力衰竭的发病机制有关。肾素作为蛋白酶,可切割血管紧张素原产生血管紧张素I。血管紧张素转换酶(ACE)是切割血管紧张素I产生血管紧张素II的关键蛋白酶,血管紧张素II(Ang II)是RAS的关键调节因子,并可通过两个G蛋白偶联受体,血管紧张素II受体1型受体(AT1R)和血管紧张素II受体2型受体(AT2R)发挥生物学功能。尽管存在其他Ang II生成酶(如组织蛋白酶和糜蛋白酶),但通常认为ACE是调节RAS中Ang II产生的关键酶,也可能是唯一有效的酶。
2000年,发现了ACE的同系物血管紧张素转换酶2(ACE2)。随后的证据表明,ACE2通过将Ang II降解为七肽血管紧张素1–7,对激活的肾素-血管紧张素系统进行负调节。一些研究支持血管紧张素1–7的反调节作用,这一作用是通过降低多数AT1受体介导的作用,特别是在血管收缩和细胞增殖方面。因此,血管紧张素1–7由于其在心血管系统中的有益作用,是RAS系统的关键组成部分。除了具有产生血管紧张素-(1–7)能力之外,ACE2是一种多功能酶,其有益效果还可能是其作用于其他血管活性肽的能力的结果。
随后,ACE2作为肽酶之外的作用逐渐得到了阐明。特别是,在2003年后,ACE2已被鉴定为非典(SARS)冠状病毒感染的一种必需受体,但也是抵抗非典致死性肺衰竭的一种保护性分子。有趣的是,ACE2的非典冠状病毒受体功能与其对Ang II降解的催化活性在机制上并无关联,而ACE2介导的Ang II降解对于肺保护免受非典型肺炎发病机制的影响仍然很重要。换句话说,SARS选择了具有作为肺保护作用的ACE2作为受体,让针对ACE2的靶向治疗(也就是上一次的假设)进退两难。
此外,ACE2及其同源物Collectrin已被鉴定为上皮细胞表面表达中性氨基酸转运蛋白所需的必需分子。Collectrin也可能在胰岛β细胞胰岛素分泌和/或胰岛细胞生长中发挥作用。
2.ACE家族分子
ACE最初在1956年被分离出来时被称为“高血压转化酶(hypertensin-converting enzyme)”。人类ACE基因位于17号染色体上,编码一种180kDa蛋白,具有两个同源结构域。每个结构域都有一个活跃的锌结合基序,His-Glu-X-X-His(HEXH基序),这种基序存在于许多肽酶中。ACE是一种I型跨膜糖蛋白,通过单个羧基末端跨膜区锚定在质膜上。在人类中,已经描述两种不同的ACE同工酶,一种是在肺内皮表面和肾、肠、胎盘和脉络丛的刷状缘膜上发现的丰富的体细胞形式,另一种是仅在睾丸中发现的ACE生发形式。这两种ACE亚型都是膜包蛋白,在细胞表面,它们作为外切酶水解循环肽。ACE可以从细胞表面裂解,从而充当可溶性酶。然而,可溶性ACE的生物学意义仍不清楚。
图1.ACE,ACE2和Collectrin的域结构
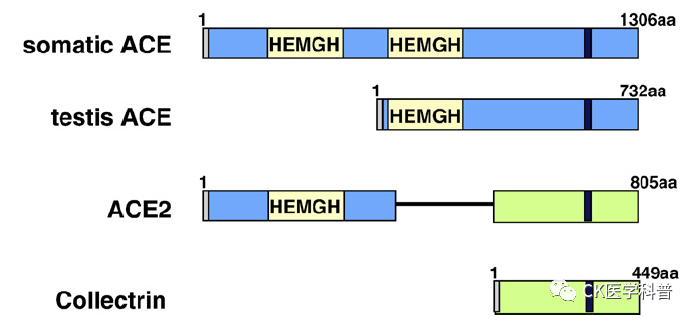
每种蛋白都是带有信号肽的I型整合蛋白,用灰色表示,而跨膜结构域则用黑色表示。锌结合基序(HEMGH)在ACE中重复两次,在ACE2中重复一次,并且位于黄色框表示的同源区域内。ACE2和Collectrin之间的同源区域以绿色表示。数字指的是每种人类蛋白质中的氨基酸数。
ACE2由805个氨基酸组成,是具有单一胞外催化结构域的I型跨膜糖蛋白。人类ACE2基因已经被克隆并被定位到X染色体上。像ACE一样,ACE2有两个结构域:氨基末端催化结构域和羧基末端结构域。催化结构域有一个活性位点--锌金属肽酶结构域--并且与ACE的氨基结构域显示出41.8%的序列一致性。ACE2的羧基末端结构域与Collectrin有48%的序列一致性,Collectrin是一种非催化蛋白,最近被证明在肾脏的氨基酸再吸收、胰腺β细胞增殖,以及可能胰岛素胞吐等方面具有关键作用。
3.ACE2功能
早期研究观察到ACE2主要在心脏、肾脏和睾丸中定位,在其他多种组织中低水平表达,尤其是结肠和肺,而后来的研究也表明ACE2在肝脏和肠等其他器官中也具有重要作用。在心脏中,ACE2在内皮细胞和心肌细胞中表达。在肾脏中,ACE2分布于管状上皮细胞的管腔表面;在睾丸中,表达于睾丸间质细胞。ACE2通常定位于上皮细胞的腔面,这与ACE相反,ACE似乎均匀分布在极化细胞的顶膜和基底外侧膜之间。而当SARS冠状病毒通过表达ACE2的细胞腔面进行感染时,其感染效力提高10倍。
3.1 ACE2的肽酶功能
ACE和ACE2都属于金属蛋白酶的M2家族,其活性位点域暴露于细胞外表面,促进循环肽的代谢。ACE和ACE2都通过利用锌催化反应,锌与活性位点内保守的组氨酸配位,促进水分子对底物羰基键的亲核攻击,形成非共价结合的中间体。除了两个组氨酸(位于HEXXH基序内),还有一个谷氨酸残基参与锌离子的配位,位于ACE和ACE2中HEXXH基序的23个氨基酸的末端。与抑制剂(MLN4760)结合的ACE2相比,天然ACE2的结构分析揭示了一个大的“铰链弯曲”运动,其中肽酶结构域的催化亚结构域I和II表现出从开放到封闭的转变。这种运动是由抑制剂的结合引起的,并为催化重新定位关键残基。
图2. ACE2在肾素-血管紧张素系统中的作用示意图
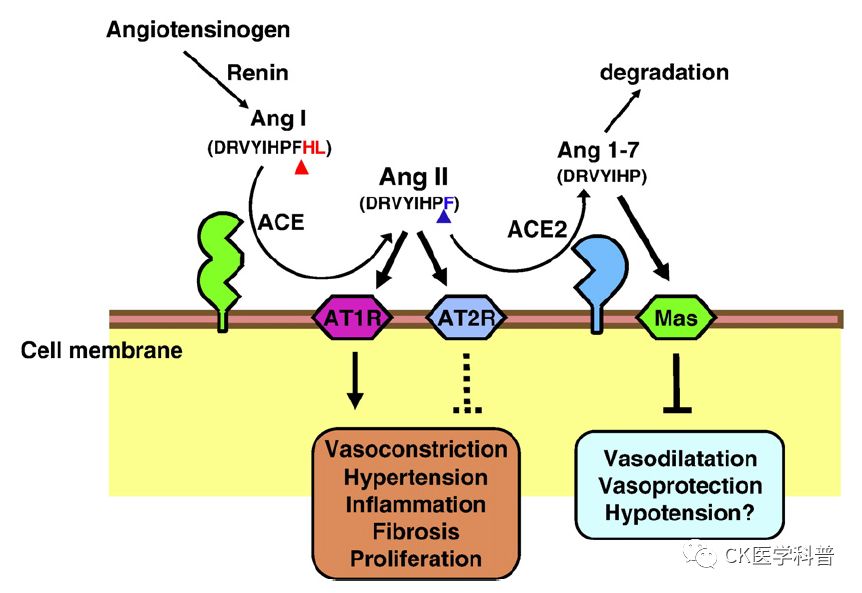
血管紧张素I(Ang I; DRVYIHPFHL)充当ACE(一种二肽基羧肽酶)的底物,并被转化为血管紧张素II(Ang II; DRVYIHPF),这是经典RAS的主要活性肽。 ACE2催化并灭活血管紧张素II,并产生血管扩张肽血管紧张素1-7(Ang 1-7; DRVYIHPF),该肽与Mas受体结合和/或降解为非活性肽。 红色箭头指示ACE裂解位点; 蓝色箭头显示ACE2裂解位点。应当指出,ACE2是一种非特异性蛋白酶,可以裂解多种其他底物,例如Apelin。
尽管有相似之处,ACE和ACE2的功能不同;ACE从其底物(二肽基肽酶,DPP)中释放一个碳端二肽,而ACE2则切割一个氨基酸(单羧肽酶)。ACE2催化可在脯氨酸和疏水或碱性碳末端残基之间优先水解的底物的肽。当AngI由ACE转化成强效血管收缩剂AngII时,ACE2可裂解Ang I,产生推测为无活性的血管紧张素1-9肽,然后可以通过ACE或其他肽酶转化为血管扩张肽Ang1-7。另外,ACE2可直接代谢Ang II产生血管紧张素1–7,其效率高于将Ang I转化为血管紧张素1–9。ACE2晶体结构的分辨率显示,这些底物特异性差异是由于精氨酸-273与底物的碳末端形成盐桥(Salt-bridger),导致ACE2中结合囊较小,而在ACE中,该残基被较小的谷氨酰胺残基取代。虽然有已知的形成Ang 1-7的酶,例如奈普利林(neprilysin)、脯氨酰内肽酶24.26和thimet寡肽酶,但ACE2的鉴定进一步支持了Ang 1-7的生物学意义。这种肽已被证明与G蛋白偶联受体Mas相互作用,介导其血管保护作用。ACE2还作用于肽Apelin-13和Apelin-36的碳末端,并在体外以高催化效率从其中切割出氨基酸。Apelin合成首先为77个氨基酸前激素,后加工成36个氨基酸肽的apelin-36;进一步蛋白水解切割产生Apelin-13。Apelin-13系统给药促进大鼠和小鼠低血压。有趣的是,Apelin-13 (F13A)的碳末端残基的修饰失去了其降压作用,并进一步拮抗野生型Apelin-13的作用,推测ACE2在Apelin肽代谢中具有作用。
ACE水解Ang I需要氯离子参与。同样,ACE2活性也受氯离子的调节。然而,氯离子存在可增加ACE2对Ang I的水解,但抑制了AngII的裂解。有人提出氯化物结合会引起活性位点构象的细微变化,这种变化会促进或阻碍底物结合。氯离子增加至超过100毫摩尔,虽然仍处于人血中生理浓度,但已可增加ACE2对Ang I的切割,减少了ACE2对AngII的切割,。这将具有增加血管收缩性的Ang II在肾脏中局部浓度的作用,此部位血管收缩性的Ang II和ACE2都有高水平的表达,且细胞外氯离子水平波动较大。
3.2 .ACE2催化活性的抑制剂和活化剂
各种ACE抑制剂,如卡托普利和赖诺普利不影响ACE2的活性,而ACE2活性可被二肽Pro-Phe抑制,并且据此已经开发了特定的ACE2抑制剂,例如肽类似物DX600和MLN 4760 ((S,S)-2-[1-羧基-2-[3- (3,5-二氯苄基)-3H-咪唑4-基]-乙胺基]-4-甲基戊酸)。MLN 4760是第一个基于Ang I的碳端二肽(His-Leu)合理设计的ACE2抑制剂,具有较高的效价(Ki=0.44 nM)和特异性。ACE2对ACE的反调节轴促使研究人员考虑ACE2对动物模型心血管疾病的可能影响。通过基因治疗或重组蛋白进行ACE2治疗确实改善了高血压、动脉粥样硬化和肾脏疾病。基于电子构象的药物筛选确定了两种ACE2激活剂化合物(xanthenone和resorcinolnaphthalein),均可中度增强ACE2活性。然而,尚不清楚这些化合物的特异性。
3.3 ACE2的肽酶非依赖性功能
尽管ACE2作为肽酶催化Ang II裂解,但最近的研究表明ACE2的跨膜区也具有生物学功能。2003年,非典疫情威胁到世界,ACE2被鉴定为致病病原体非典冠状病毒的功能受体。表达ACE2非催化活性突变体的细胞仍然允许非典病毒感染,这表明ACE2的肽酶作用对于非典病毒进入宿主细胞不是必需的。与生物学结果相一致,结构分析表明,非典冠状病毒Spike蛋白接触ACE2催化结构域的亚结构域I的顶端,但不影响亚结构域II,也不封闭肽酶活性位点。当非典型肺炎冠状病毒与ACE2连接时,ACE2的外结构域被裂解,而跨膜结构域被内在化,使病毒颗粒-宿主细胞进一步融合。因此,尽管详细的机制仍不清楚,但ACE2的跨膜区与非典冠状病毒-受体复合物在非典冠状病毒感染中从细胞膜到细胞质的转运有关。
图3. ACE2的翻译后修饰; 内化和脱落

SARS冠状病毒(SARS-CoV)以Clathrin蛋白依赖性方式与ACE2结合并内在化,以使其进入细胞。 膜融合是通过蛋白酶(例如胰蛋白酶或furin蛋白酶)Spike介导激活,病毒RNA被释放到细胞质中,从而引发SARS感染。 跨膜蛋白酶(ADAM17)切割ACE2的细胞外近膜区域,将催化活性的胞外域释放到细胞外环境中。 尚不清楚这种ACE2裂解是否有助于SARS发病。
图4. ACE2与B0AT1氨基酸转运蛋白的相互作用

ACE2与B0AT1氨基酸转运蛋白(SLC6A19)相互作用,这是肠道上皮细胞中该转运蛋白的极化表面表达所必需的。 尚不清楚ACE2的切割是否有助于为B0AT1提供中性氨基酸。
大鼠肾脏分离的Collectrin基因在再生收集管中的表达分析。Collectrin与ACE2的碳末端有47.8%的同一性;然而,与ACE2不同,Collectrin缺乏活性羧肽酶催化结构域(图1)。初次报告记录了Collectrin定位在集合管上皮细胞的细胞质中,但进一步的研究表明Collectrin主要定位在近端管状上皮细胞的刷状缘(管腔侧)。通过对小鼠的基因定位研究,偶然发现Collectrin是中性氨基酸转运蛋白的重要调节因子。Collectrin敲除小鼠的尿液中出现过量的中性氨基酸(酪氨酸和苯丙氨酸)。生化研究表明,Collectrin与B0AT1中性氨基酸转运蛋白结合,并对这些转运蛋白在肾近端小管氨基酸再吸收所需的细胞表面的正确表达起关键作用。尽管结构相似,ACE2并不与肾脏中的氨基酸转运蛋白结合,而是与肠道中的氨基酸转运蛋白结合,在肠道中ACE2高度表达,氨基酸被吸收。而ACE2的这一功能与其肽酶活性无关,其肽酶活性不是与氨基酸转运蛋白配对所必需。
图1.ACE,ACE2和Collectrin的域结构
每种蛋白都是带有信号肽的I型整合蛋白,用灰色表示,而跨膜结构域则用黑色表示。锌结合基序(HEMGH)在ACE中重复两次,在ACE2中重复一次,并且位于黄色框表示的同源区域内。ACE2和Collectrin之间的同源区域以绿色表示。数字指的是每种人类蛋白质中的氨基酸数。
4.ACE2表达的调节
4.1 .ACE2的转录调控
ACE2最初是使用人类衰竭性心室的cDNA文库克隆的,而ACE2 mRNA水平的表达则根据生理和病理条件而动态变化。目前越来越多的证据表明,ACE抑制剂或AT1受体阻滞剂对RAS的抑制作用会上调ACE2mRNA的表达。抑制盐皮质激素(或醛固酮)可能通过抑制氧化应激而增加了巨噬细胞中的ACE2 mRNA。包括Ang II、细胞因子和NF-κB在内的炎症信号可能会抑制ACE2转录。干扰素-γ和白细胞介素-4下调上皮细胞中ACE2基因的表达。因此,炎症信号,包括Ang II、细胞因子和核因子κB,均可能抑制ACE2转录。
Ace2敲除小鼠心脏缺氧诱导基因的上调。组织局部缺氧增加了人和大鼠心肌梗死中ACE2的表达但在大鼠模型研究中,没有观察到心肌梗死中ACE2基因水平的变化。ACE2过度表达抑制心脏成纤维细胞缺氧诱导的胶原生成。在缺氧的肺平滑肌细胞中,缺氧早期的ACE2基因水平升高,HIF(缺氧诱导因子)-1α积累后的后期降低至接近基线水平。因此,低氧条件下ACE2表达的调控仍然难以明确,可能是环境或细胞/器官依赖性的。全反式维甲酸也显示出能提高自发性高血压大鼠的ACE2基因水平。肝细胞核因子1β (HNF-1β,TCF2)学内分泌的都应该大致了解,是一种组织特异性转录因子,其在人类中的突变可能会导致肾囊肿、生殖器畸形、胰腺萎缩和MODY5。在细胞系中,ACE2被鉴定为HNF-1β的直接靶基因,并非HNF-1α (TCF1),并且在ACE2启动子区有多个HNF-1β结合位点。ACE2同源物Collectrin位于靠近X染色体上的ACE2位点,也是HNF-1转录因子的靶基因,包括胰腺β细胞中的HNF-1α和肾上皮细胞中的HNF-1β。因此,我们可以推测ACE2和Collectrin基因的表达是由HNF-1转录因子协同调节的。
4.2 .ACE2脱落和内化
ACE2被鉴定为非典冠状病毒受体,据报道,ACE2作为完整分子和/或其跨膜区在感染时与非典病毒外壳一起被内化,此内吞作用对病毒感染至关重要。即使重组SARS表面配体 Spike蛋白与ACE2相互作用时,内化也能发生。已经有人提出两种途径,即Clathrin蛋白依赖性和非依赖性非典型肺炎冠状病毒进入靶细胞途径。然而,ACE2细胞质尾的作用是有争议的;例如在另一项研究中,ACE2细胞质尾的缺失并不影响非典型肺炎-CoV的进入,但它会减弱这一过程。与ACE相似,ACE2可受到近膜裂解事件(脱落)的影响,释放催化活性胞外结构域。佛波酯、离子霉素、内毒素、白细胞介素-1β或肿瘤坏死因子α可刺激该过程。脱落是由混杂的“sheddase”,ADAM17(或TACE,肿瘤坏死因子-α转化酶;图3)介导,ADAM17-敲除细胞中,ACE2脱落减少。此外,钙调蛋白结合位点在ACE2的胞质尾部被鉴定,钙调蛋白的抑制增加ACE2胞外结构域向培养上清液的释放(脱落)。尽管因为循环ACE2和残留的胞内结构域的作用尚未确定,因为ACE2胞外结构域脱落的生理作用仍然难以确定,但脱落似乎与非典型肺炎-CoV细胞的进入和复制有关,并且ADAM17抑制剂可在体外抑制非典型肺炎-CoV的复制。
参考文献:
-
http://engine.scichina.com/publisher/scp/journal/SCLS/doi/10.1007/s11427-020-1637-5?slug=abstract
-
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(20)30183-5/fulltext
-
https://www.sciencedirect.com/science/article/abs/pii/S0163725810001415?via%3Dihub
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#ACE#
48
#ACE2#
47
学习了
132