Nat Med发表迄今最大、最全面的人肺细胞图谱,加速未来肺部单细胞研究
2023-07-07 测序中国 测序中国 发表于上海

研究团队整合了已发表的和新生成的数据集,构建了一个完整的人类呼吸系统综合单细胞转录组图谱——人类肺细胞图谱。
导读
过去十年间,单细胞技术的快速发展使得单细胞数据集在规模和数量上都得到了增长,但一般研究通常只能捕获有限的供体,并在细胞类型的定义上存在一定的分歧。因此,整合多种单细胞数据集能够解决单一研究的这些局限性,同时也能捕获群体中存在的变异性、以及个体间细胞图谱的多样性。
整合的单细胞图谱提供了在单一研究中无法获得的新见解,最近的参考图谱也发现了未知的细胞类型,鉴定了在研究中可重复的标记基因等。但许多目前可用的图谱在人类样本数量、每个器官的细胞类型以及供体的基础信息数据方面受到限制,这导致其无法代表并分类健康器官内和个体间细胞表型的多样性。
此外,在整合不同来源的数据时,将技术偏差与生物学相关信息正确分离至关重要,但大多数现有数据集尚未评估其数据集成的质量。因此,将现有的数据集成功地整合到一个单一的组织图谱中是实现人类细胞图谱目标的关键步骤。
近日,德国亥姆霍兹慕尼黑研究中心计算生物学研究所、慕尼黑工业大学等机构的研究团队合作在Nature Medicine发表了题为“An integrated cell atlas of the lung in health and disease”的文章。研究团队整合了已发表的和新生成的数据集,构建了一个完整的人类呼吸系统(包括上呼吸道和下呼吸道)综合单细胞转录组图谱——人类肺细胞图谱(HLCA)。HLCA包含来自486个供体和49个数据集的数据,共涵盖240万个细胞,其提供了一个具有匹配标记基因的共识细胞类型重新注释,包括稀有和先前未被描述的细胞类型。此外,该研究还通过HLCA发现了多种肺部疾病的共同细胞状态,有助于确定新的治疗靶点。HLCA扩展了人们对健康肺及其疾病变化的理解,可作为未来分析肺数据的参考。

文章发表在Nature Medicine
主要研究内容
数据集成构建HLCA
研究团队使用先进的机器学习技术,汇集、整合了近40项主要单细胞RNA测序人类肺部研究的49个数据集,创建了第一个整合的人类肺细胞图谱HLCA。该图谱的核心是一个完全整合的14个健康参考数据集,共涉及61种细胞身份,包含罕见和新颖的细胞类型,并具有对人类肺细胞图谱的数据衍生的共识注释。除健康肺部数据外,研究团队还从10多种不同的肺部疾病中获取数据集,并将这些数据投射到健康数据上,以了解疾病状态。
为实现有监督的数据集成和下游集成分析,研究团队开发了一个五级层次细胞身份参考框架来协调细胞类型命名。将数据生成器提供的每个数据集的细胞身份标签映射到分层参考框架,以统一数据集的细胞类型标记,并显示每个样本中不同的细胞类型比例。
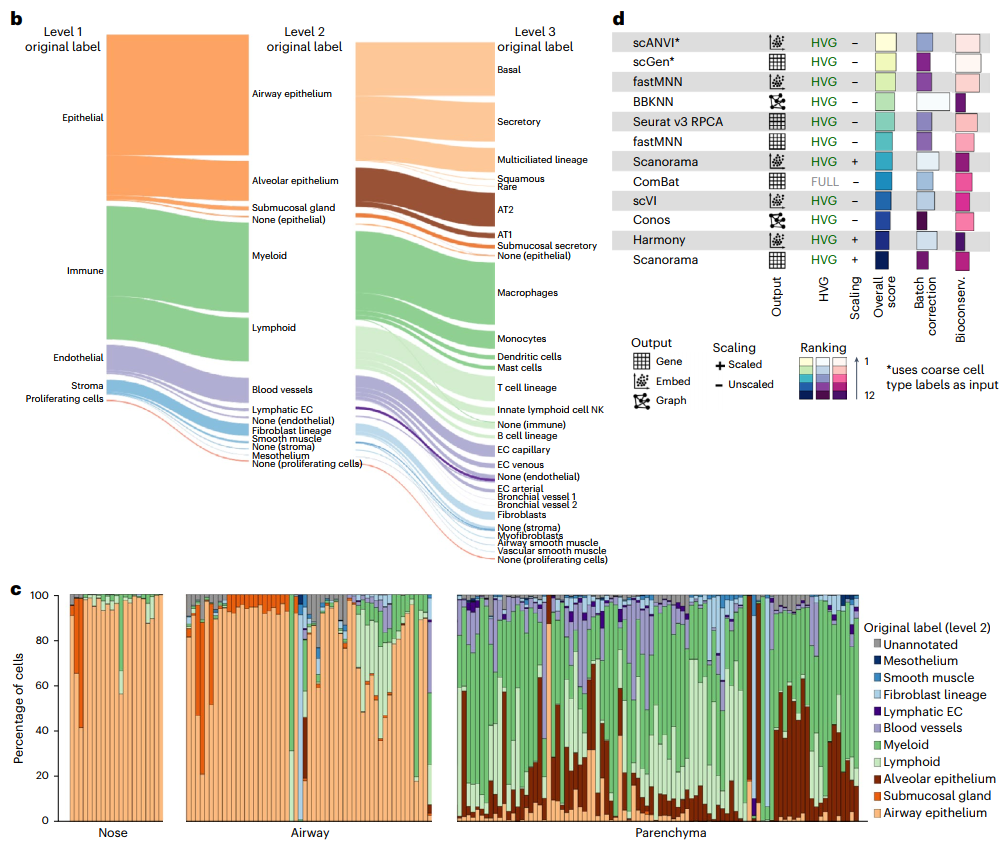
图1. HLCA的组成
HLCA可识别罕见及新的细胞类型
为确定不同数据集中一致和不一致的区域,研究团队迭代地对HLCA核心进行聚类,并使用熵值评分检测聚类中的供体多样性和细胞类型标签一致性。大多数聚类显示出较低的标签熵,这表明数据集之间的细胞类型标签总体上是一致的;有33个亚群表现出高标签熵,表明其识别了错误标记的细胞类型,需要对集成图谱进行一致的重新注释。
作为对HLCA核心中存在的细胞类型多样性进行共识的第一步,研究团队在原始注释和六个专家意见的基础上对集成数据进行了全面的重新注释。结果显示,共识细胞类型注释部分有41%对应于原始标签、28%存在改进、31%进行了大量重新注释。为了稳健地表征细胞类型,研究团队还建立了一套通用的标记基因,可应用在不同研究中。此外,该研究还发现了6种新的肺部细胞类型。
综上,完全重新注释的HLCA结合了来自不同研究的数据,为健康肺组织中的细胞类型注释和标记基因提供了精细的参考。HLCA能够改进稀有细胞类型的检测和鉴定、发现未知细胞类型。
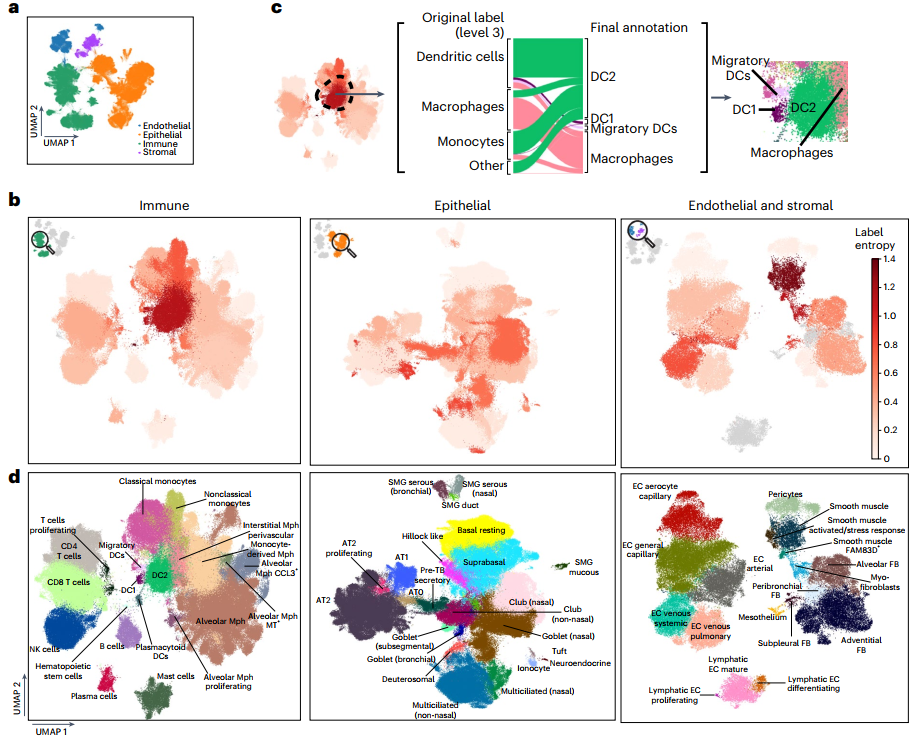
图2. HLCA支持共识驱动的细胞注释
供体和实验因素影响基因表达谱
人口统计学和其他元数据协变量(如年龄、体重指数BMI等)可影响细胞转录表型,研究团队对其进行了分析,以分析每种技术或协变量对细胞类型特异性基因表达变异的影响。结果显示,解剖位置是影响样本间大部分差异的生物学变量;性别与淋巴内皮细胞的转录组变异相关性最强,BMI与B细胞和T细胞的变异最相关。此外,对于几种细胞类型(如肥大细胞、AT1细胞等),组织分离方案揭示了所记录技术和生物协变量的大部分差异。
为更好地描述生物变量如何影响细胞表型,研究团队还在基因水平上模拟了其对转录组的细胞类型特异性影响。结果显示,淋巴内皮细胞中性别相关的差异主要由位于X和Y染色体上的基因表达差异主导;细胞类型特异性程序随呼吸道近端到远端的位置而变化。随着BMI的增加,供体中几种细胞类型显示出转录组学变化:AT2细胞、分泌细胞和肺泡巨噬细胞表现出一系列生物过程的下调。上述结果表明,通过HLCA可详细了解解剖和人口统计学协变量对肺细胞图谱的影响及其与疾病的关系。
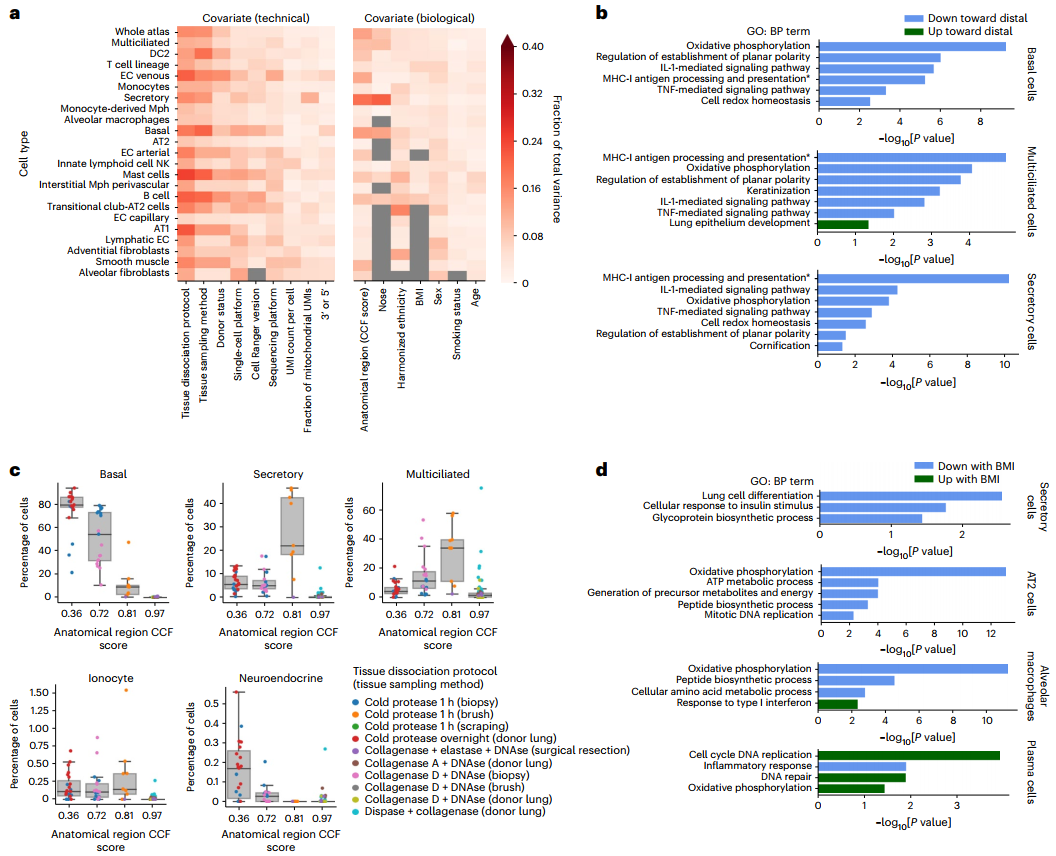
图3. 驱动个体间差异的人口统计学和技术变量分析
将数据映射到HLCA中可显示与疾病相关的状态
为扩展HLCA图谱,研究团队使用scArches将来自37个数据集的380名健康/患病个体的细胞映射到HLCA中。经平均标签转移不确定性评分评估,有27个数据集与HLCA映射良好,其余10个数据集来自COVID-19研究。在这些数据集中,较高的不确定性值可能由映射数据与HLCA成人健康肺样本之间存在的真正生物学差异所致。总体而言,成功映射的数据集包括疾病样本以及来自多种状态下的单细胞数据,显示出HLCA作为通用参考的潜力。
肺部疾病的特点是出现独特的与疾病相关的转录表型。研究团队观察到病变肺样本数据集的标签转移不确定性更高,表明标记细胞类型可能因疾病而改变。具体而言,在特发性肺纤维化(IPFs)中,肺泡成纤维细胞和肺泡巨噬细胞的标签相互作用形成失调的细胞回路,与来自相同数据的健康对照样本相比,IPF样本中的标签转移具有更高的不确定性。因此,HLCA可用于注释新数据,识别以前未报告的群体,使用标签转移不确定性评分可帮助检测疾病影响的细胞状态和相应的基因表达程序。
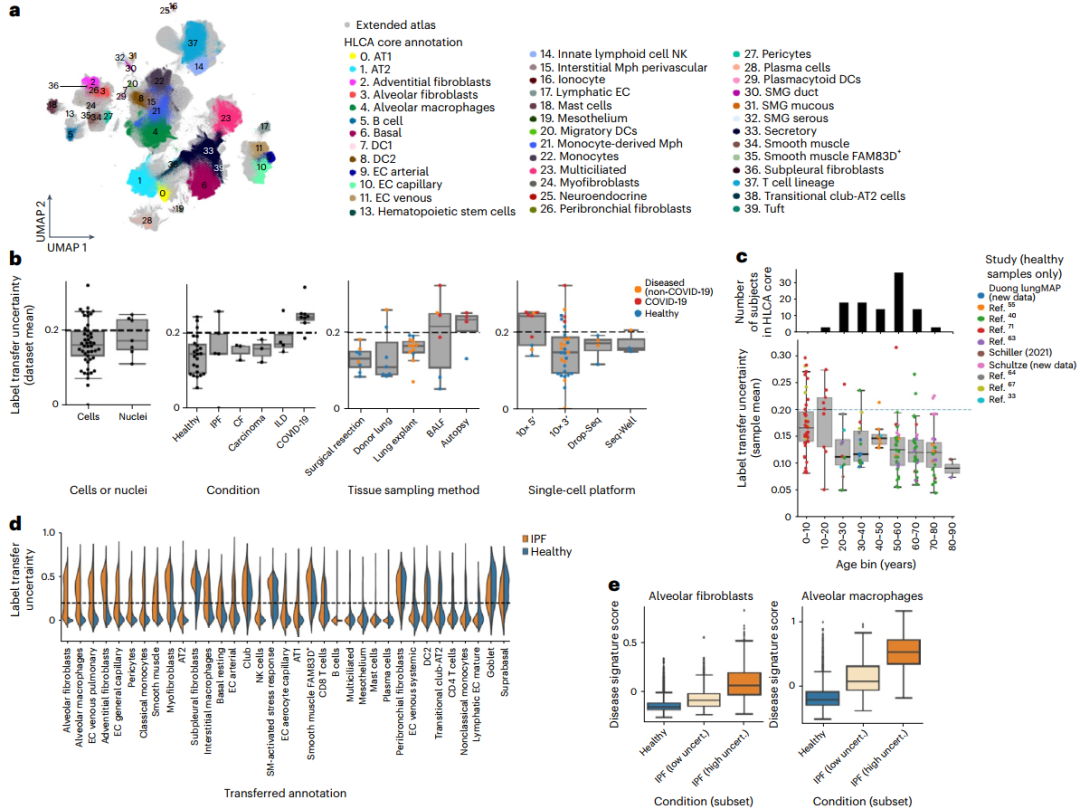
图4. 扩展的HLCA能够识别疾病相关的细胞状态
结 语
综上所述,研究团队建立了人类呼吸系统的综合参考图谱。HLCA整合了来自49个数据集的数据,产生了240万个细胞的参考,涵盖了迄今为止发表的所有主要肺scRNA-seq研究。通过将数据投射到HLCA上,研究团队发现HLCA能够快速详细地注释新数据集,识别独特、与疾病相关的细胞状态和多种疾病共有的细胞状态,如肺部纤维化、癌症和COVID-19等。总之,HLCA可作为肺单细胞相关研究的普遍参考,为更好、更全面地了解人类肺部的健康和疾病铺平了道路。
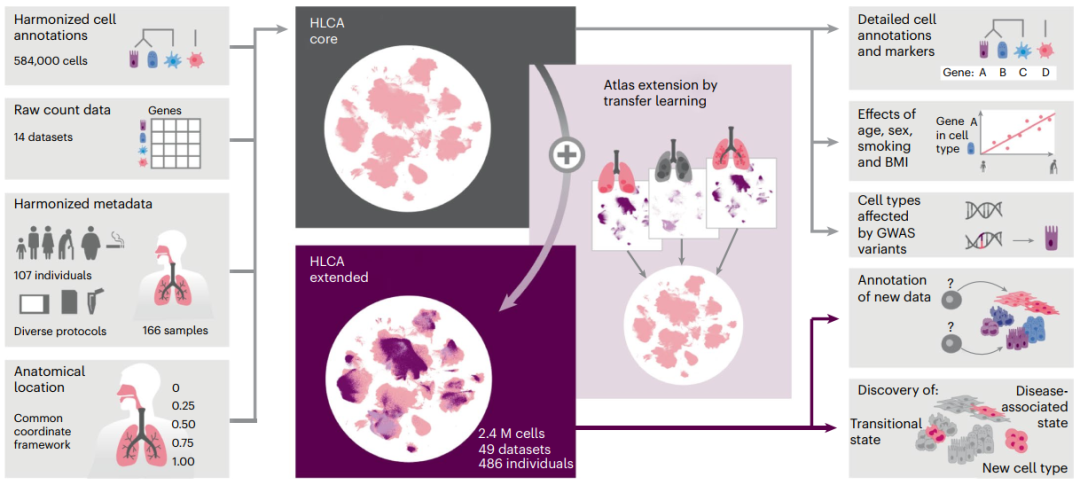
研究概括图
参考文献
1. Sikkema, L., Ramírez-Suástegui, C., Strobl, D.C. et al. An integrated cell atlas of the lung in health and disease. Nat Med (2023).
2. Steuernagel, L. et al. HypoMap—a unified single-cell gene expression atlas of the murine hypothalamus. Nat. Metab. 4, 1402–1419 (2022).
3. Salcher, S. et al. High-resolution single-cell atlas reveals diversity and plasticity of tissue-resident neutrophils in non-small cell lung cancer. Cancer Cell 40, 1503–1520.e8 (2022).
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



