
先天性镜像运动障碍(CMM)| 疑难探究
2023-09-17 神经科学论坛 神经科学论坛 发表于上海

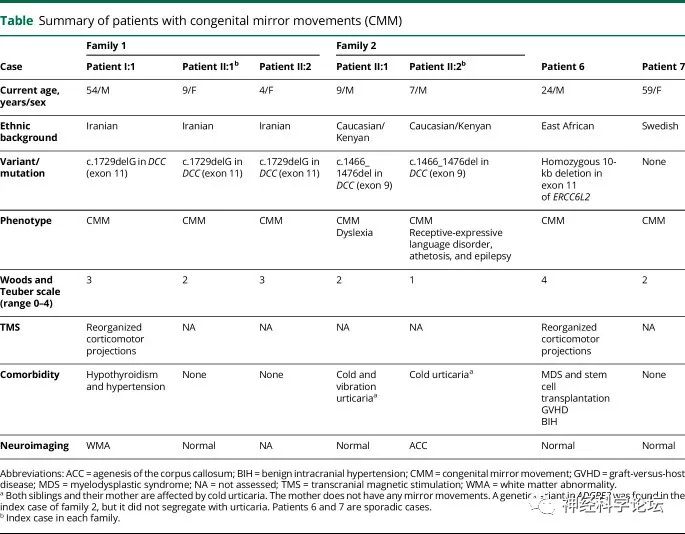
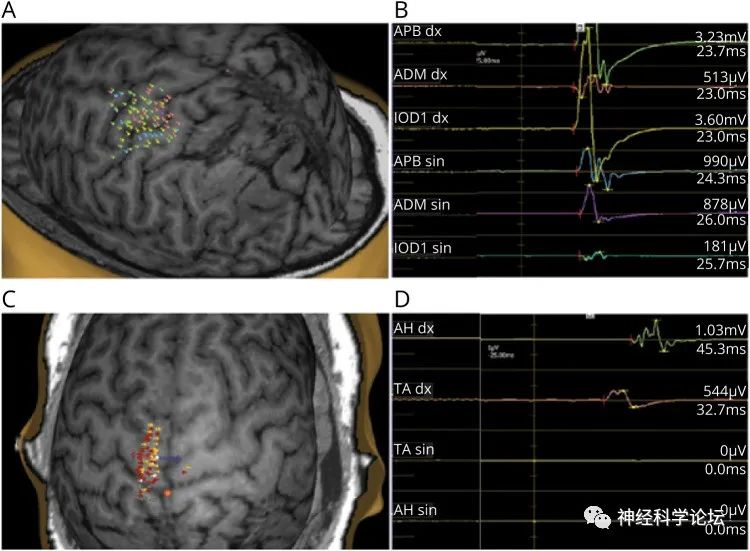
CMM与皮质脊髓束的异常交叉有关,皮质脊髓束是自主运动的主要运动束。已知RAD51在同源重组中起关键作用,在DNA修复中具有关键功能。虽然首次提出RAD51单倍充分性来解释CMM,但也可能涉及其他机制
论坛导读:镜像运动是一只手的无意识运动,反映了另一只手的有意运动。先天性镜像运动(CMM)是一种罕见的常染色体显性遗传疾病,镜像运动是其主要的神经系统表现。CMM与皮质脊髓束的异常交叉有关,皮质脊髓束是自主运动的主要运动束。已知RAD51在同源重组中起关键作用,在DNA修复中具有关键功能。虽然首次提出RAD51单倍充分性来解释CMM,但也可能涉及其他机制。
镜像运动也可以发生在没有先天性镜像运动障碍的人身上。温和的镜像运动在儿童正常发育期间很常见,通常在7岁之前消失。他们也可以在晚年生活在患有神经退行性疾病如帕金森症的人身上。先天性镜像运动障碍(congenital mirror movements,CMM)的特点是在通常没有其他临床体征或症状的个体中,出现早发、明显的镜像运动(身体一侧的不自主运动,反映对侧的有意运动)。患有CMM的人在日常生活的某些方面会有一些困难,特别是那些需要每只手做不同动作的活动,例如在键盘上打字。此外较长时间使用手时,他们会明显感到上肢不适甚至疼痛。虽然镜像运动的严重程度不同,但大多数受影响的个体具有比相应的自主运动幅度更小的强烈和持续的镜像运动。镜像运动通常持续一生,没有恶化或改善,并且通常与随后出现的其他神经症状无关。其临床特征是早发性的显著的镜像性动作,即一侧肢体作随意运动时,另侧肢体同时出现同源性肌肉的不随意运动。然而,DCC中杂合致病变异的受影响个体子集可能患有CMM,伴有胼胝体异常和伴随的认知和/或神经精神问题。神经心理障碍的严重程度与镜像运动和/或胼胝体发育不全有关。分离的DCC致病变异体与最佳预后相关。
CMM较罕见,估计患病率低于1:1,000,000。一般以常染色体显性(AD)方式遗传,大多数患有由DCC、NTN1或RAD51中的致病性变异引起的CMM的个体从可能有症状或无症状的父母那里遗传了致病性变异。如果先证者的双亲之一受到影响和/或具有DCC、NTN1或RAD51致病性变异体,其同胞继承该变异体的风险为50%。值得注意的是,先证者的亲本临床上未受影响的同胞患CMM的风险仍然增加,因为杂合子亲本外显率降低的可能性很大。患有AD CMM的个体的每个孩子有50%的机会继承致病变体;然而,由于外显率降低,遗传致病性变异的后代可能不会出现CMM。一旦在受影响的家庭成员中识别出引起CMM的致病变异体,就有可能进行产前和植入前基因检测。
临床特征:
-
镜像运动的严重程度各异,但运动幅度小于相应的随意运动。常用的描述术语为联带运动(synkinesis)、双手协同运动(bimanual synergia)。
-
患者的日常活动存在中等度困难,包括不能执行纯粹的单手运动以及完成需要熟练双手协调的任务,偶尔会在持续手工活动期间出现上肢疼痛。
-
CMM没有感觉异常,但少数患者可表现为感觉耦合(一侧肢体可感受到对侧肢体接受的刺激)。
-
镜像运动通常持续终生,但不会改善或恶化,也通常不伴有后续的其他神经系统表现,但一些患者可共病认知和/或神经精神问题。
-
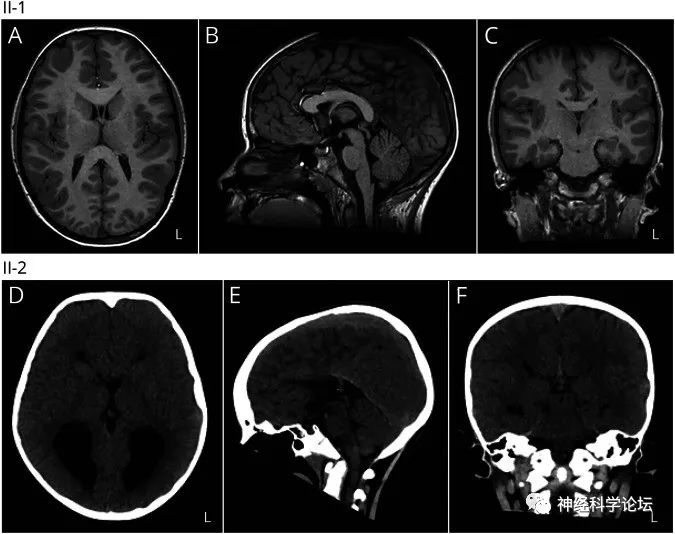
大部分患者头MRI正常,但一些DCC基因杂合致病突变的患者可表现胼胝体部分或完全发育不良。
在镜像表型分类中最常用的神经心理学测试早于更详细的表型分型技术的发展,而不仅仅依赖于主观评级。仅基于视觉观察的结论甚至可能对是否进行特定的神经心理学测试产生偏见;当可能存在细微效应(亚临床镜像3)时,这也可能导致“无镜像”的分类。虽然由于一系列不可避免的因素(如患者的局限性、可用性、时间),特定的神经心理学测试可能无法在一些个体中实施,但所包括的亚组中报告的测量结果的不一致性导致难以得出明确的结论。生理性镜像运动指正常发育的幼儿可出现轻度的生理性镜像运动,通常在7岁前完全消失,但易在老年时逐渐复发。但早发型镜像运动综合征(Syndromes with early-onset mirror movements)并不总是孤立存在的,可以是一些复杂的综合征和先天性轻偏瘫的一个组成部分。需要鉴别的综合征包括Kallmann 综合征 (KS) (ANOS1基因突变,嗅觉减退和低促性腺素性功能减退症)、Joubert 综合征(小脑蚓部发育不良,伴特征性的磨牙征及不同的神经症状)、Klippel-Feil 综合征 (KFS) (颈椎先天融合,后发际低,短颈和颈部运动幅度下降)、Moebius syndrome(先天性非进行性面部无力,单眼或双眼外展)等。
CMM是一组非渐进性运动障碍,对侧肢体在有意运动中出现异常镜像。CMM在儿童期表现为主要累及上肢,持续一生,通常在没有其他神经症状的情况下观察到。DDC netrin 1受体(DCC)、RAD51和netrin-1 (NTN1)的致病变异与CMM相关。到目前为止,只有1个同源的CMM大家族被报道在动力蛋白轴突轻链4 (DNAL4)中含有纯合变体4;一些CMM病例/家族缺乏可检测的突变。DCC突变与大脑和脊髓连合连接中断引起的一系列神经系统综合征相关。DCC编码一种netrin-1受体,该受体参与穿过中线的发育轴突导向,这可能是异常同侧皮质投射的原因。netrin-1受体由4个细胞外免疫球蛋白样(Ig1-4)、6个纤连蛋白3样(FN1-6)结构域、一个跨膜结构域和3个细胞内结构域(P1-3)组成。DCC中的杂合子致病变异体与孤立镜像运动(MMs)、胼胝体发育不全(ACC)、伤害感受性位置认知的异常发展和皮质脊髓束(CST)的重组投射相关,无论是孤立现象还是组合现象。双等位基因DCC突变导致严重的发育性裂脑综合征。
与先天性镜像运动相关的脑畸形有:父亲和女儿的胼胝体发育不良伴DCC杂合子p.Met1*突变;TUBB3杂合子p.Thr312Met突变的母亲和儿子的胼胝体发育不良、脑回障碍和畸形蚓部;TUBB基因p.Arg121Trp杂合子突变的一个父亲和两个女儿的胼胝体发育不良、脑回运动障碍、蚓部异常和脑室不对称;TUBA1A中p.Glu155Asp杂合子突变患者的胼胝体发育不良、脑回障碍、基底神经节畸形和蚓部异常;POMGNT1中p.Pro312Leu纯合子突变患者的脑积水、发育不全的胼胝体、多微回和小脑囊肿。
CMM的诊断是有相关症状的先证者,经基因鉴定发现DCC、NTN1或RAD51的杂合致病突变。具有镜像运动持续存在于整个成年期,缺乏其他提示某种综合征的临床证据和后续的神经系统发现的临床特征、头颅MRI正常或者存在胼胝体部分或完全发育不良的影像发现以及家族史的个体要怀疑CMM。
参考文献
Nissenkorn A, Yosovich K, Leibovitz Z, Hartman TG, Zelcer I, Hugirat M, Lev D, Lerman-Sagie T, Blumkin L. Congenital Mirror Movements Associated With Brain Malformations. J Child Neurol. 2021 Jun;36(7):545-555. doi: 10.1177/0883073820984068.
Trouillard O, Dupaigne P, Dunoyer M, Doulazmi M, Herlin MK, Frismand S, Riou A, Legros V, Chevreux G, Veaute X, Busso D, Fouquet C, Saint-Martin C, Méneret A, Trembleau A, Dusart I, Dubacq C, Roze E. Congenital mirror movements are associated with defective polymerisation of RAD51. J Med Genet. 2023 Jun 12:jmg-2023-109189. doi: 10.1136/jmg-2023-109189.
Thams S, Islam M, Lindefeldt M, Nordgren A, Granberg T, Tesi B, Barbany G, Nilsson D, Paucar M. Heterozygous variants in DCC: Beyond congenital mirror movements. Neurol Genet. 2020 Oct 20;6(6):e526. doi: 10.1212/NXG.0000000000000526.
Franz EA. Characterizing the phenotypes of congenital mirror movements and other rare genetic disorders. Dev Med Child Neurol. 2020 Jun;62(6):669. doi: 10.1111/dmcn.14509.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言



