可变性红斑角皮症 1 例
2018-05-21 张莉 刘彤云 临床皮肤科杂志

患者男,22岁。因掌跖增厚20余年,全身反复鳞屑性红斑伴瘙痒5年,于2012年7月12日来本院就诊。
【一般资料】
患者男,22岁。
【主诉】
因掌跖增厚20余年,全身反复鳞屑性红斑伴瘙痒5年,于2012年7月12日来本院就诊。
【现病史】
患者出生后不久双手掌、足跖皮肤增厚,表面附有薄层鳞屑,继而出现线状皲裂,未诊治。5年前患者自述无明显诱因于躯干及四肢出现大小不等片状暗褐色斑疹,散在分布,表面粗糙,可见鳞屑。皮损逐渐增多,部分融合成片状角化过度性斑片,并逐渐波及腋窝、胸背部、脐周、臀部及四肢,伴瘙痒。遇冷后瘙痒可缓解,夏季严重,冬季减轻,但病情反复,近年来皮损瘙痒加剧,曾多次于外院就诊,诊断为“湿疹”,具体治疗不详,病情无改善。
【个人史】
既往体健,父母非近亲结婚,否认家族中有类似疾病史。
【体格检查】
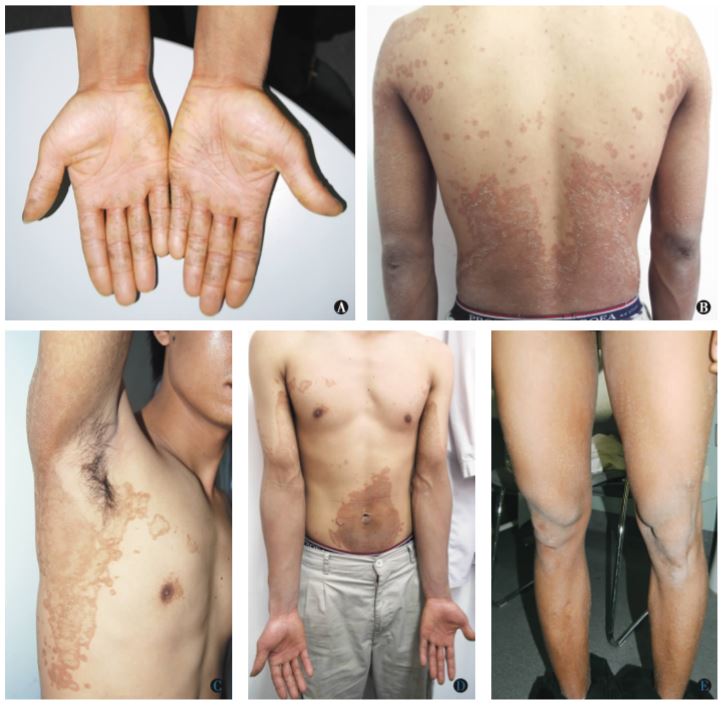
系统检査无异常。皮肤科检查:躯干、脐周、双腋下及四肢可见对称分布大小不等的片状、环状及地图状暗褐色角化性斑片及斑块,边界清楚,表面粗糙,上覆薄层糠状鳞屑,散在分布绿豆至黄豆大角化过度性丘疹及斑丘疹,双手掌及跖部皮肤增厚、粗糙,见浅表线状皲裂纹(图1),毛发、牙齿及指(趾)甲无异常。
【辅助检查】
实验室及辅助检查:血、尿、粪常规,肝、肾功能,血糖,电解质,血脂及心肌酶谱均正常。抗人类免疫缺陷病毒(HIV)抗体、抗核抗体(ANA)、抗可溶性抗原(ENA)抗体均正常或阴性。胸部X线片正常,B超示:肝、胆、脾、胰、肾、膀胱及前列腺均正常。皮损组织病理检查:表皮角化过度伴有角化不全,颗粒层存在,棘层不规则肥厚,轻度乳头瘤样增生,基底细胞散在空泡变性,表皮突不规则向下延伸;真皮浅层毛细血管增生、扩张,血管周围稀疏的淋巴细胞浸润(图2)。
【初步诊断】
可变性红斑角皮症。
【治疗】
予系统使用阿维A20mg/d口服,局部外用地奈德乳膏及阿达帕林凝胶。患者于第2周复诊,全身红斑明显变淡、变暗,瘙痒缓解,腋下红斑区中央部分消退呈环形。
【讨论】
可变性红斑角皮症(erythrokeratodermiavari-abilis)是一种少见遗传病,其遗传方式可能为不规则常染色体显性遗传,但也有常染色体隐性遗传的报道,致病基因一般认为是编码连接蛋白的GJB3或GJB4基因及其亚家族,但也有一些个案报道中未发现连接蛋白异常。本病的发病机制不明,可能因系统性外胚层血管发育不良和异常的血管扩张扰乱表皮细胞角化,电子显微镜检查可在颗粒层与角质层看见大量的谷粒细胞,可见密集分布的张力微丝,在红斑区或正常皮肤可见无髓鞘神经轴突和施万细胞,这些神经和细胞的分布可能使皮损随着温度及情绪的变化而变化。本病多为幼年发病,儿童期加重,部分患者出生时便有皮损。皮损可持续、反复发作,并加剧至成年,青春期后减轻持续终身不愈,但一般不影响健康。本病临床表现因人而异,主要的皮损特点为形态各异、边界清楚的角化过度性红斑及斑块,主要有两型:①有固定形态的角化过度性斑块,在正常皮肤或红斑基础上的散在、持久性红棕色角化过度性斑片或斑块,边界清楚,状如地图;②可自行消退的红斑损害,其大小、数量、形态及位置因气候或情绪变化可在数小时、数日或数周变化明显。本病分布多发生于四肢伸侧、臀部及面部,也可发生于全身各个部位。常无自觉症状,或轻度瘙痒。本病组织病理无特异性,角化过度,乳头瘤样增生,颗粒层增厚以及真皮血管扩张和单核样细胞浸润。本例患者出生2年后发病,青春期加重,病情反复,临床特征为边界清楚的角化过度性斑片、斑块及角化过度性丘疹及斑丘疹。根据患者病史、临床特点及组织病理学表现,诊断为可变性红斑角皮症。本病需与进行性对称性红斑角化病及毛发红糠疹等病相鉴别。本病一线治疗主要是局部外用药物,包括角质剥脱剂、维A酸、他扎罗汀、α-羟基酸及糖皮质激素等。二线用药主要是系统使用依曲替酯、阿维A或异维A酸。维A酸类药物联合补骨脂素长波紫外线(UVA)照射有很好的疗效。本例患者系统使用阿维A、局部外用地奈德乳膏及阿达帕林凝胶后第2周复诊,全身红斑明显变淡、变暗,瘙痒缓解,病情明显好转。
原始出处:
张莉, 刘彤云. 可变性红斑角皮症1例[J]. 临床皮肤科杂志, 2017(6):442-444.
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#红斑#
18
#变性#
24