
Nature Genetics:重度抑郁症的多祖先全基因组关联研究有助于发现位点、精细映射、基因优先排序和因果推断
2024-02-19 Jenny Ou MedSci原创 发表于上海

这些发现表明,对于医学博士来说,在遗传研究中增加祖先和全球多样性对于确保发现核心基因和告知发现的可转移性可能特别重要。
重度抑郁症(MD)是最紧迫的全球健康挑战之一。虽然全基因组关联研究(GWAS)有望揭示MD发展背后的生物机制,但它们揭示了一种高度多基因遗传结构,其特征是变异,单独导致小风险增加,可能是由于MD症状和病因的异质性。之前的遗传研究探讨了不同结果定义、性别和创伤暴露对异质性的影响。然而,血统和种族在MD遗传学中的作用尚未得到系统评估。

到目前为止,MD的GWAS主要在欧洲血统中进行。最大的MD GWAS结合了几项研究的数据,并确定了223个独立的显著单核苷酸多态性(SNP)。该研究还包括来自百万退伍军人计划(MVP)队列的59,600名非裔美国人的数据。在他们的双祖先荟萃分析中,重要的SNP数量增加到233个。其他MD GWAS在样本量有限的非裔美国人和西班牙裔/拉丁美洲参与者中进行,没有发现与MD有统计学意义关联的变体。
CONVERGE研究有10,640名中国女性参与者,是迄今为止在“西方”国家以外进行的最大的MD GWAS。该研究确定了两个与线粒体生物学相关的全基因组重要关联,并报告了欧洲祖先样本中与MD的遗传相关性为0.3318。与此相一致,我们最近的工作表明,在欧洲祖先样本中进行的一些先前确定的GWAS位点不能转移到东亚祖先样本。
在评估MD风险因素的因果效应时,遗传效应的异质性可能会影响发现。之前对欧洲祖先样本的研究报告了MD和心脏代谢结果之间的遗传相关性和因果关系。值得注意的是,我们之前的研究表明,东亚个人和欧洲血统个人的MD与体重指数(BMI)之间的联系方向相互矛盾(BMI的积极因果效应)。因此,使用门德尔随机化(MR)调查不同祖先群体和不同疾病亚型的因果关系,对于确保可推广性和区分风险因素与疾病之间关系的生物和社会机制非常重要。
增加基因研究的多样性对于确保公平的健康益处也很重要。在美国,不同种族群体在MD的表述上的差异可能会影响诊断的可能性。为欧洲血统参与者优化的遗传学将主要使该组患者受益,因此可以进一步扩大各组之间的诊断和治疗差异。
2024年1月4日发表在Nature Genetics的文章中,使用了来自不同祖先的样本的数据,并进行了全基因组关联元分析,然后对目标基因进行精细映射和优先排序。我们评估了遗传基因座在祖先群体之间的可转移性。最后,我们探讨了MD和心脏代谢特征之间的双向因果关系。
在这里,我们报告了MD的多祖先GWAS,将21个队列的数据与之前报告的数据相加,其中有88,316个MD病例和902,757个对照组。该分析使用一系列措施来定义MD,并包括非洲(有效样本量的36%)、东亚(26%)和南亚(6%)血统以及西班牙裔/拉丁美洲参与者(32%)的样本。
研究结果显示,多祖先GWAS确定了53个显著相关的新位点。对于欧洲祖先样本中GWAS的位点,可转移到其他祖先群体的位点比预期的要少。精细映射受益于额外的样本多样性。一项全转录组关联研究确定了205个显著相关的新基因。
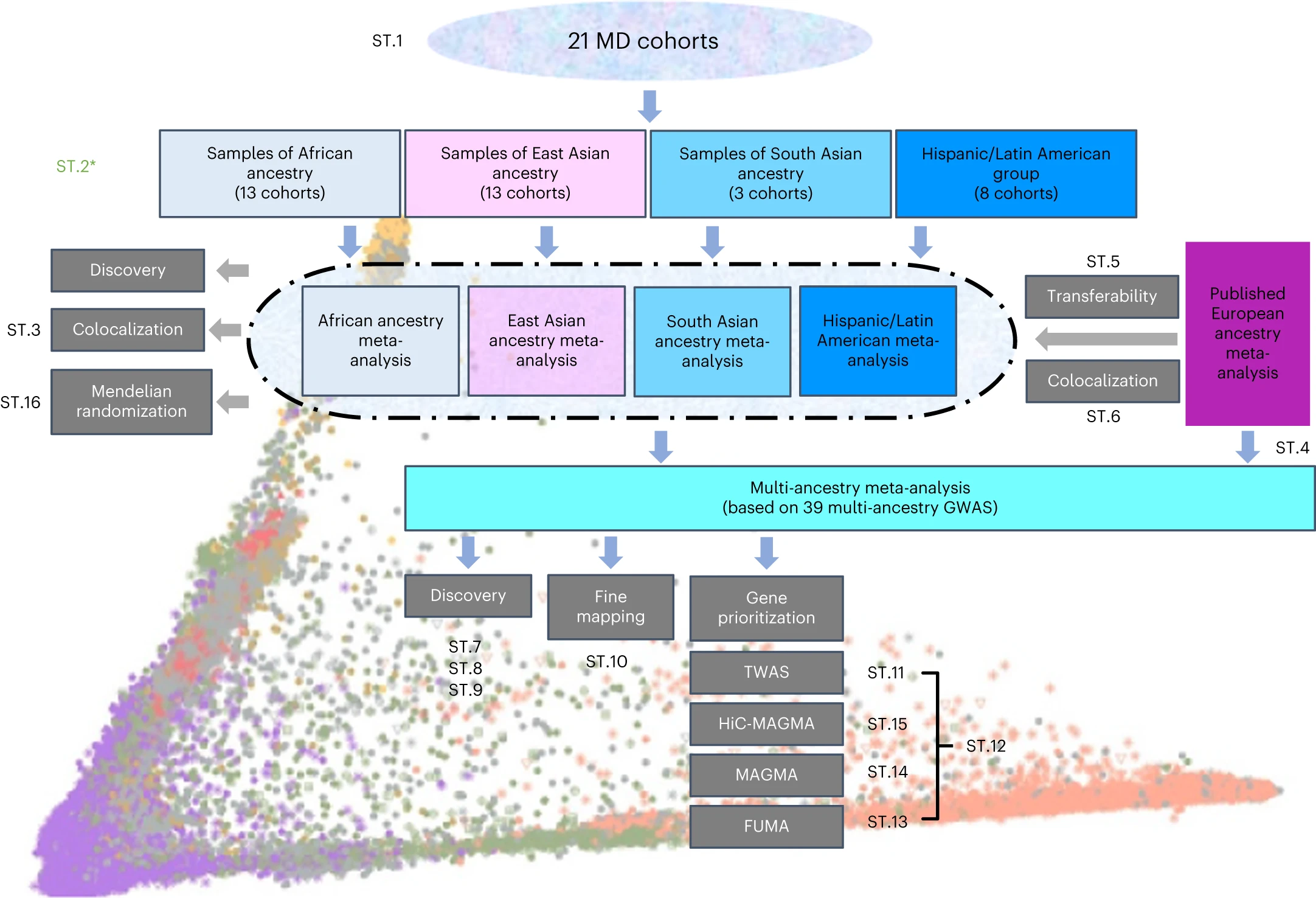
本研究分析的示意图
综上所述,在MD的第一个大规模多祖先GWAS中,我们通过可转移性分析证明了MD位点的显著比例是欧洲祖先样本特有的。我们确定了欧洲血统分析中遗漏的新型、生物上似是而非的关联,并证明了大量、多样化的样本对于识别目标基因和假定机制非常重要。这些发现表明,对于MD来说,一种具有高度复杂病因的异质性条件,遗传研究中增加祖先和全球多样性可能对于确保发现核心基因并告知不同祖先群体的发现的可转移性尤为重要。
原文出处
Meng, X., Navoly, G., Giannakopoulou, O. et al. Multi-ancestry genome-wide association study of major depression aids locus discovery, fine mapping, gene prioritization and causal inference. Nat Genet 56, 222–233 (2024). https://doi.org/10.1038/s41588-023-01596-4
 s41588-023-01596-4.pdf
s41588-023-01596-4.pdf
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言











#全基因组# #重度抑郁症#
26