
神经棘细胞增多症(NA):表现为咬舌型共济失调|病例分享
2023-06-19 神经科学论坛 神经科学论坛 发表于上海

本病主要缺陷是血中β脂蛋白减少或缺乏,根据遗传方式Kell血型,分为常染色体隐性或显性遗传的舞蹈病-棘形红细胞增多症与X-连锁Mcleod综合征两种类型,其特征为进行性神经退行性变。
论坛导读:神经棘红细胞增多症(neuroacanthocytosis,NA )是一种罕见的遗传性的脂类代谢病,又称为Bassen-Kornzweig综合征,无β-脂蛋白血症(betaripoproteinemia)或棘红细胞增多症。本病主要缺陷是血中β脂蛋白减少或缺乏,1960年Levine等首次描述此病,根据遗传方式Kell血型,分为常染色体隐性或显性遗传的舞蹈病-棘形红细胞增多症(chorea-acanthocytosisCA)与X-连锁Mcleod综合征两种类型,其特征为进行性神经退行性变,伴舞蹈样动作及棘形红细胞增多。
病例1
32岁女性听力受损,出现意识丧失、癫痫发作和咬舌。由于听力损失,有交流困难。她需要气管插管来防止血液阻塞气道。头部CT显示没有萎缩的双侧尾状核和侧脑室扩大。在住院后观察到不自主的口周运动。第八天癫痫发作。再次进行气管插管和机械通气,以防止不自主的口周运动和咬舌。她表明她不需要舌头,说明她患有精神疾病。几年来,她去过许多医院,并用了抗惊厥药,但没有明确诊断出躯体疾病。
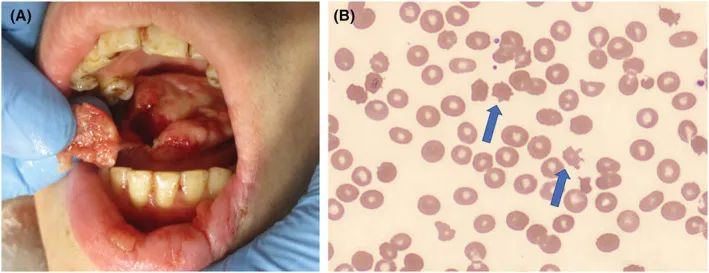
图源:Acute Med Surg. 2023 Jan 16;10(1):e815.
由于自残、不自主的口周运动、躯干共济失调、癫痫史以及亲属中只有她的兄弟表现出的类似症状而出现的舌和唇畸形,提示棘红细胞增多症,且在外周血涂片上鉴定出棘红细胞。
根据年龄、家族史、使用的药物、血液检查和影像学检查结果,确定并排除了有症状的舞蹈病、药物诱发的舞蹈病、代谢性疾病和其他神经退行性疾病。后对她的不自主运动进行了深部脑刺激手术。手术后,她的不自主运动改善,步态稳定,咬舌消失。尽管精神障碍可能表现为自残和疑似癫痫,但不自主运动应提示考虑遗传性疾病。
病例2
59岁女性,不自主舌运动影响进食和说话1年。她最初于2016年10月出现症状。该患者还有1年的焦虑、不安和认知障碍史。精神病医生诊断她是强迫症,并提供心理治疗。随后,她的症状出现波动。在接下来的3个月里,病人的高动力不自主运动变得越来越明显。她出现了舌突出、口面运动障碍、构音障碍、吞咽困难、全身性舞蹈病、足部张力障碍性运动、双侧反射消失和张力减退,但没有明显的肌无力和肌萎缩。患者体温正常,既往病史不明显。Babinski、Chaddock和脑膜刺激征呈阴性。
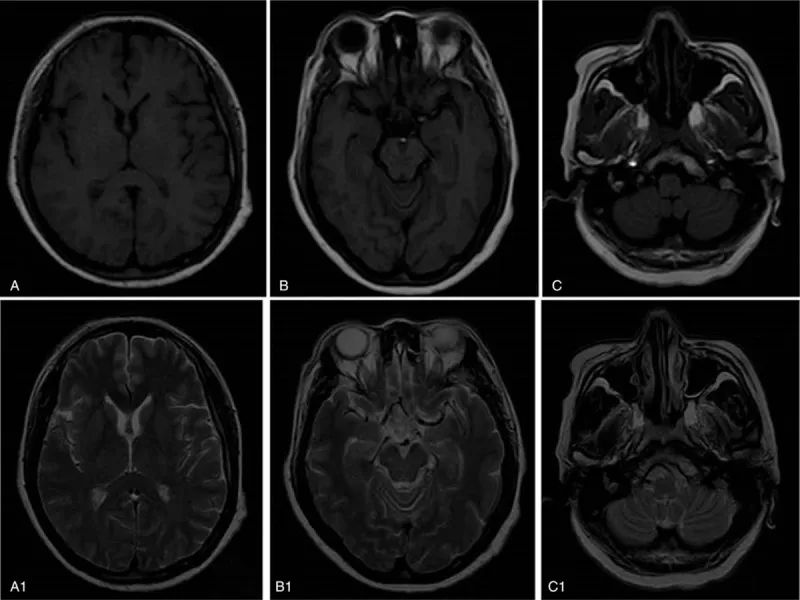
图源: Medicine (Baltimore). 2019 Jan;98(2):e14050.
脑磁共振成像(MRI)显示双侧苍白球、额叶、颞叶和小脑轻度年龄相关性萎缩。脑电图和脑脊液分析正常。血清肌酸激酶(CK)水平为612U/L(正常范围为25–200 u/L)。血常规分析、血沉、肝功能、肾功能、甲状腺功能以及血清叶酸、维生素B12和肿瘤标志物水平正常。患者对抗HIV抗体和梅毒没有反应。在外周血涂片中观察到棘红细胞。

图源: Medicine (Baltimore). 2019 Jan;98(2):e14050.
症状包括舌突出、口面运动障碍、构音障碍、全身性舞蹈病、足部张力障碍性运动、反射消失、无肌病、认知障碍、神经精神障碍、血清CK水平升高和外周血涂片中的棘红细胞被认为符合ChAc的诊断。进行了基因检测,在XK、PANK2、APOB、SC5D、KCNN4、MTTP、VPS13A或GATA1基因中没有发现突变。患者口服硫必利(100毫克,每日三次)治疗舞蹈病,阿普唑仑(0.8毫克,睡前每日一次)改善睡眠,维生素B1 (20毫克,每日三次)和维生素B12 (0.05毫克,每日三次)。经过2个月的治疗,患者的症状明显减轻。在6个月的随访中,患者可以自己进食,并在没有帮助的情况下行走。
延伸阅读
神经棘细胞增多症(NA)是一种异质性遗传性神经退行性疾病,以畸形的针状红细胞(棘细胞)和类似亨廷顿舞蹈病的症状为特征。NA综合征表现为运动障碍,如舞蹈症、口面运动障碍、肌张力障碍和帕金森综合征,以及认知障碍、精神症状、癫痫发作和轴突神经病。NA的病理生理学涉及基底神经节的进行性变性。NA综合征很罕见,估计患病率为1-5/100万。NA多见于青春期或成年早期,发病年龄8~62岁;病程7~24年,存活最长者达33年;男性多于女性,男女之比约为1.8∶1。核心NA综合征包括由液泡蛋白分类13同源物A (VPS13A)基因突变引起的舞蹈病-棘红细胞增多症(ChAc ),以及由Kx基因突变引起的麦克劳德综合征(MLS)。可能伴有棘红细胞增多症的其他遗传性神经退行性疾病包括亨廷顿病样2 (HDL2)、泛酸激酶相关神经变性(PKAN)和伴有脑铁积聚的神经变性(NBIA)。NA综合征的鉴别诊断基于其基因型和表型异质性。
神经棘红细胞增多症(NA)是由Yamamoto等人于1982年首先用于描述神经系统疾病和棘红细胞同时发生的组合。随着时间的推移,这一概念不断发展,目前核心NAC综合征包括舞蹈病‐棘红细胞增多症(ChAc)、麦克劳德综合征、亨廷顿舞蹈病(HD)样和泛酸激酶相关的神经变性。Hardie等人三十年前报道的最大的NAC综合征系列包括19个病例。多年来,术语“神经棘细胞增多症”(neuroacanthocytosis, NA)的使用充满了分类学上的混乱。这种持续的混淆主要是由于NA已经在各种不同的分类水平上被用于指代神经系统疾病,在这些疾病中,变形的“尖状”红细胞(棘红细胞;在其他情况下也称为毛刺单元)。最广义地说,NA是指脂质吸收障碍(如Bassen-Kornzweig病),其中存在外周神经病和小脑体征,以及基底神经节变性障碍,其特征为运动障碍。前一组疾病现在通常更恰当地定义为它们的代谢或遗传异常(例如分别为无脂蛋白血症或MTTP突变)。
在更具体的层面上,在近20年前定义的NA是一个总括术语,指的是第二组疾病。其中,将两种疾病称为“核心”NA综合征,舞蹈病-棘红细胞增多症(ChAc常染色体隐性遗传,OMIM #200150)和麦克劳德综合征(X连锁隐性遗传,OMIM #300842),将它们与另外两种疾病分组,亨廷顿病样2(HDL 2;常染色体显性,OMIM #606438)和泛酸激酶相关的神经变性(PKAN常染色体隐性遗传,OMIM #606157),其中也有棘红细胞增多症的报道。在第三个水平上,NA也是,并且继续是,用作ChAc的同义词,指由于VPS13A的双等位基因突变而患有特定神经退行性疾病的患者。
舞蹈病-棘红细胞增多症(ChAc)是一种罕见的常染色体隐性神经退行性疾病,由空泡蛋白分类13A (VPS13A)的致病变异体引起。迄今为止,只有少数ChAc患者被报道,VPS13A的变异谱尚未完全阐明。文献报道过一36岁女性,她从30岁开始就经历了口面运动障碍,使用二代测序的遗传研究中,鉴定出VPS13A的2种变体,无义变体c.4411C>T (p.Arg1471Ter)和剪接变体c.145-2A>T。通过文献回顾,剪接变异体c.145-2A>T被新归类为致病变异体。因此,根据典型的临床表现、实验室检查结果和影像学结果,该患者被诊断为ChAc。
意大利报告的一个相对较大的NA患者队列研究,证明ChAc和MLS虽然罕见,但在疑似遗传性舞蹈病的鉴别诊断中应始终考虑。根据可用的实验室技术,可以首先测试具有蛋白质印迹和Kell血型表达的chorein剂量,或者进行VPS13A和XK基因的DNA测序。此外,由于现在怀疑原发性肌病不仅存在于MLS患者中,也存在于ChAc患者中,因此建议对所有确诊的NA患者进行肌肉活检和蛋白表达研究。
参考文献
Kim A, Chae HY, Park HS. Compound Heterozygous VPS13A Variants in a Patient with Neuroacanthocytosis: A Case Report and Review of the Literature. Lab Med. 2022 Jul 4;53(4):433-435.
Sogabe T, Ishida K, Ogawa H, Ohnishi M. Tongue-biting ataxia that appeared to be a psychiatric disorder: a case of neuroacanthocytosis. Acute Med Surg. 2023 Jan 16;10(1):e815.
Walker RH, Danek A. "Neuroacanthocytosis" - Overdue for a Taxonomic Update. Tremor Other Hyperkinet Mov (N Y). 2021 Jan 11;11:1.
Vaisfeld A, et al. Neuroacanthocytosis Syndromes in an Italian Cohort: Clinical Spectrum, High Genetic Variability and Muscle Involvement. Genes (Basel). 2021 Feb 26;12(3):344.
Zhu H, Feng XM, Zhao T, Liu JY. Neuroacanthocytosis with unusual clinical features: A case report. Medicine (Baltimore). 2019 Jan;98(2):e14050.
Neeraja K, et al. The Spectrum of Movement Disorders in Neuroacanthocytosis Syndromes: A Video Series. Mov Disord Clin Pract. 2021 Jul 1;8(6):983-986.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言




进修期间见过这种病例,基层医院诊断这个太难
32