
肝豆状核变性:Leipzig评分诊断系统
2023-07-28 神经科学论坛 神经科学论坛 发表于上海

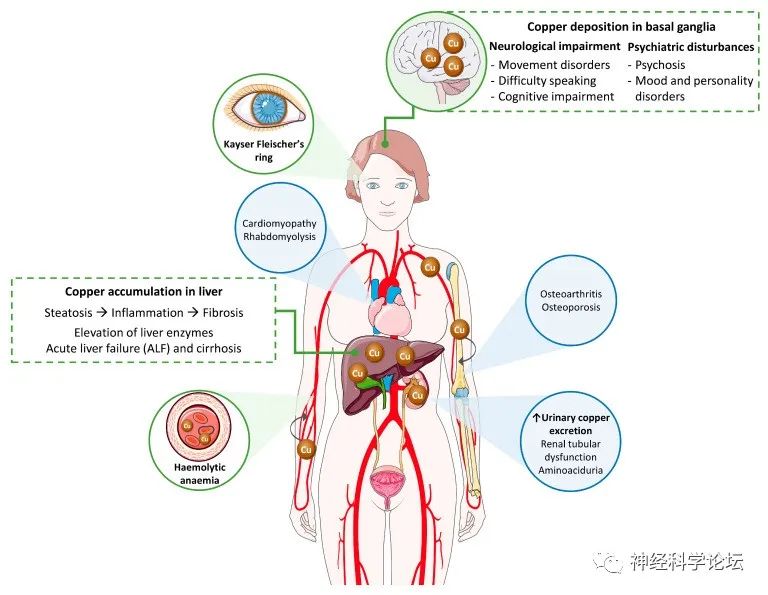
肝豆状核变性是一种常染色体隐性遗传病,其病变基础为编码铜跨膜转运蛋白的基因ATP7B发生突变,导致铜在肝脏、大脑和其他组织器官中过度沉积,从而引起各种临床表现,其中最常见的是进行性肝病和神经精神障碍。
论坛导读:Wilson病(WD)又称为肝豆状核变性,是一种常染色体隐性遗传病,其病变基础为编码铜跨膜转运蛋白的基因ATP7B发生突变,导致铜在肝脏、大脑和其他组织器官中过度沉积,从而引起各种临床表现,其中最常见的是进行性肝病和神经精神障碍。WD的发病报道年龄从2~78岁不等,但主要以儿童及青少年多见。WD是一种多系统疾病,症状多样,以铜的异常沉积为特征。临床症状取决于病理过程中铜积累的器官。
因此,肝脏和神经系统受累占优势,WD可以表现为肝脏、神经系统或混合疾病。大多数儿科患者通常表现为肝脏表现,而成年患者主要表现为混合表现。虽然诊断通常在儿童期、青春期或成年早期,即5至35岁之间做出,但也有晚期表现的描述。
据统计,世界范围内WD患病率为1/5000~1/30 000,ATP7B突变基因携带率为1/90,但存在地区差异。WD是可用药物治疗的遗传代谢性疾病,其长期预后取决于治疗的早晚。治疗越早,损害越轻,预后越好。WD一经确诊,即应尽快开始药物治疗。约80%~85%接受治疗的WD患者长期预后良好。但WD患者对药物反应个体差异大,目前还没有适合所有WD患者的治疗药物,应根据患者情况选择适当的治疗方案。抗铜治疗不能纠正患者的基因缺陷,即使治疗效果良好,也不能终止治疗,停药会导致病情反复、恶化、甚至肝衰竭。虽然WD的诊断指南已经制定,但患者的识别仍然是一个挑战,这导致了不可逆的严重并发症的延迟诊断和发展,如永久性脑损伤或肝损伤,这使得治疗无效。
WD的主要特征是肝脏疾病、神经精神异常和KF环。具有神经系统表现的KF环和/或低血清铜蓝蛋白(Cp)的存在被认为足以确定WD的诊断。然而,大多数病例需要结合临床症状和实验室评估。目前,没有一种单一的检测方法可以对每一个潜在的患者进行新的WD诊断。WD杂合子携带者偶尔会出现Cp水平异常降低。对低CP水平的其他考虑是吸收不良、暴发性肝衰竭或Cp突变引起的无血清血症。因此,Cp作为诊断WD的独特标记物的预测价值是值得怀疑的,主要是因为许多WD患者显示出边缘水平。在最近对Cochrane数据库的系统综述中,发现阈值0.20 g/L的敏感性为77.1-99%,特异性为55.9-82.8%,而值< 0.1 g/L的敏感性为65-78.9%,特异性为96.6-100%。神经性WD患者的血清Cp降低,但在多达50%的患有活动性肝病的成年患者中,血清Cp可处于低正常范围内,并且血清Cp对WD诊断的阳性预测值很低。在WD儿童中,15~36%的Cp在正常范围内。单独的血清Cp不足以诊断或排除WD。
虽然临床分子诊断的最新进展大大提高了受影响患者及其同胞的WD诊断的准确性,但传统的Sanger测序不能检测大的缺失或重复。此外,在ATP7B基因中有许多单核苷酸多态性(SNPs)和未知意义的变体(VUS)。基因测序结果的解释,特别是在只有一个确定的突变或VUS存在的情况下,造成WD患者识别的模糊性。
目前提出了一种诊断评分-Leipzig评分系统2003来指导临床诊断,并且已经在2012欧洲肝脏研究协会的临床实践指南中采用。Leipzig评分系统全面系统地纳入了WD的临床表现、生化检验和遗传分子学检测结果,内容包括KF环的有无、神经系统症状的有无或严重程度、血清铜蓝蛋白水平、Coomb’s实验阴性的溶血性贫血有无、肝铜定量及肝组织罗丹宁染色结果、24 h尿铜水平、ATP7B基因检测结果,每项按照有无或程度不同设1~4分不等;每项内容的分数相加,当Leipzig总分≥4分时,即可诊断为WD。
WD诊断的Leipzig评分系统
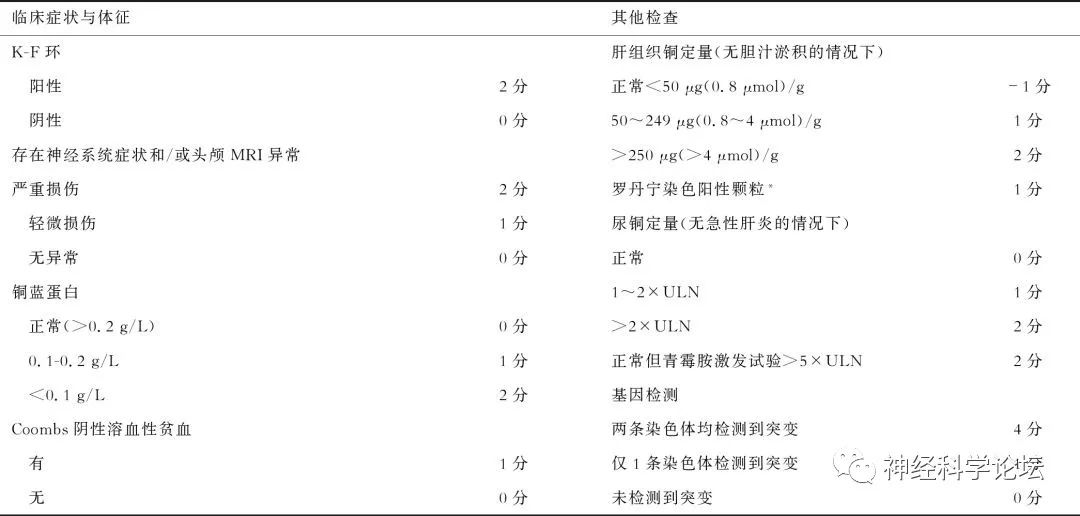
总分≥4分可确诊;总分3分为疑似诊断,需进一步检查;总分≤2分基本不考虑诊断。注:*如不能进行肝铜定量时采用;ULN:正常值上限。
欧洲肝病学会WD临床实践指南推荐的Leipzig评分系统是目前应用最广泛的诊断方法,该评分系统凸显基因诊断的重要地位。当患者ATP7B基因存在2个致病性突变(复合杂合致病突变或纯合致病突变)时,按照Leipzig评分可得4分,即可诊断为WD。然而,在临床实践中,考虑到WD常常需要终身药物治疗,而青霉胺等药物常具有不同程度的毒副作用,对于无症状患者,是否仅凭借基因检测就可以确诊WD并立即启动临床治疗,尚存在不同声音,甚至造成一定的临床决策困惑,值得深入探讨。
需要指出的是,根据Leipzig评分系统患者两条染色体上均检出ATP7B致病突变时分值为4分,故WD是少数经基因检测就可以进行临床诊断的遗传代谢性疾病。既往也有过病例报道,对存在H1069Q纯合子突变的WD先证者的同胞,未给予其任何系统治疗,在诊断WD 50年后也未出现任何症状,有学者认为上述案例的发生可能与WD复杂的发病机制有关。尽管如此,目前Leipzig评分仍被普遍推荐作为WD的诊断标准。根据Leipzig评分系统患者评分≥4分可确诊,3分为可以诊断。但如果WD患者症状可不典型,血清铜蓝蛋白可不降低,可无K-F环,尿铜可不升高,肝组织铜染色也可阴性,临床患者诊断应综合患者各项指标进行评价,尤其注意进行ATP7B基因检测。
目前,ATP7B基因检测是协助WD诊断的重要手段。然而,研究显示,ATP7B基因外显率并不是100%。部分人群即使携带ATP7B纯合或复合杂合致病突变,体内也不一定存在铜代谢障碍,不见得发病。许多基因突变可能只是条件致病性的,只有当遗传和外部环境等上述因素间相互作用,使患者体内铜蓄积到某种概念上的阈值时,才会产生病理状态。因此,对于无症状WD患者,分子诊断仍具有挑战性,仅凭基因检测有时并不足以做出临床和治疗决策,尚需结合其他检查(如铜蓝蛋白、24小时尿铜定量、眼睛KF环、肝功能、肝组织铜定量、肝脏及头颅影像学等)来综合判断。部分仅检出存在纯合或复合杂合致病性突变、而无任何症状及体征的人群,经过全面评估,可以在患者充分知情同意的情况下,可暂不进行治疗,但应定期进行随访监测。
为了获得合理的诊断成功率,建议进行分层遗传分析,目的是覆盖整个ATP7B序列和不同的可能突变类型。首先,建议进行21个编码外显子和侧翼内含子序列的研究,因为它们包含大多数(~95%)临床变异。这种方法可以通过Sanger测序来完成,Sanger测序允许检测点突变以及小的缺失和插入。在少数病理变化占主导地位的人群中,优先考虑这些变化(可能是创始人事件)是一种经济有效的策略。为此,可以使用HRM(高分辨率熔解)技术、定制的SNP(单核苷酸多态性)微阵列或RFLPs(限制性片段长度多态性)测试。
现有的临床标准和基因检测对肝豆状核变性(WD)的诊断都有很大的局限性,经常造成患者识别的模糊性,导致延误诊断和无效治疗。通过直接测量患者干血斑样本中的替代肽来指示ATP7B蛋白浓度,可以提供WD的主要证据。ATP7B肽的定量有效地识别了92.1%的WD患者,减少了铜蓝蛋白和基因分析的不确定性。基因结果不明确的患者可以获得清晰的结果,这大大有助于无创性诊断。结合ATP7B肽浓度的建议诊断评分和算法可以快速诊断和补充当前的Leipzig评分系统。即使是基于Leipzig评分的初始WD诊断的患者,也可能存在模仿WD表型(Wilson样)的其他情况。许多患者使用当前可用的方法进行诊断,但其他人仍然处于不确定的领域,因为接近铜蓝蛋白水平,不确定的遗传发现和不清楚的表型。目前,可用于WD的生物标志物是肝脏或24小时尿液中的铜蓝蛋白和铜,但它们还不够可靠。因此,生物标志物的表征使能够预测疾病的发展,并监测新药,这对改善其诊断和预后至关重要。
对于WD患者有多种诊断、治疗和监测工具。虽然许多患者通过目前可用的方法得到了诊断和充分评估,但诊断和监测已显示出对陷入诊断不确定性的其他患者更具挑战性,所述诊断不确定性的特征在于临界铜蓝蛋白水平、不确定的遗传发现和不清楚的临床表型。患者预后基于早期诊断和适当的长期治疗。由于延迟诊断因此延迟开始除铜治疗会导致灾难性的后果。遗传学测试、神经成像、生物标志物表征和先进疗法方面的技术进步可能为未来几年更准确地检测和管理WD提供信心。
参考文献
Collins CJ, et al. Direct Measurement of ATP7B Peptides Is Highly Effective in the Diagnosis of Wilson Disease. Gastroenterology. 2021 Jun;160(7):2367-2382.e1.
Ryan A, et al. Biomarkers for diagnosis of Wilson's disease. Cochrane Database Syst Rev. 2019 Nov 19;2019(11):CD012267.
梁晨, 郑素军. ATP7B基因外显率降低的机制及对Wilson病诊断的影响[J]. 肝脏, 2020, 25(10): 1123-1125.
Xu A, et al. Development and evaluation of an unlabeled probe high-resolution melting assay for detection of ATP7B mutations in Wilson's disease. J Clin Lab Anal. 2017 Jul;31(4):e22064.
Sánchez-Monteagudo A, et al. Wilson's Disease: Facing the Challenge of Diagnosing a Rare Disease. Biomedicines. 2021 Aug 28;9(9):1100.
梁晨, 白丽, 郑素军. Wilson病基因型-表型关系、诊断、治疗及筛查研究进展[J]. 临床肝胆病杂志, 2019, 35(9): 2116-2119.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言







学习WD的评分系统
33