选择性PARP7抑制剂研发进展
2023-01-16 精准药物 精准药物 发表于安徽省

近期PARP7被报道为抑制IFN-I信号传导以响应胞质NA传感器配体和几种类型的RNA病毒的主要AHR调控基因。
走进PARP领域下一个明星靶点PARP7
PARP领域一直是人们研究的热点领域。在2014年,FDA批准了首个获批的 PARP抑制剂 —奥拉帕利,PARP抑制剂是首款根据合成致死原理研发的抗癌药,目前全球已有6款PARP抑制剂药物获批问世,2020年全球市场规模已超过31.35亿美元,根据Evaluate Pharma的预测,到2023年PARP抑制剂的销售额合计将达到45亿美元。
PARP酶家族的成员使用β-NAD+作为底物连续将ADP-核糖部分添加到靶蛋白上(PARsylation)催化蛋白翻译后修饰。PARsylation除了在DNA修复中得到充分研究的作用外,还被证明可以调节多种过程,包括细胞增殖,DNA甲基化,细胞凋亡,转录调节和WNT信号传导。
PARP家族部分基于催化活性可分为三类:monoPARPS催化单ADP-核糖单元转移到其底物上,包括大多数PARP家族成员;polyPARPS催化poly-ADP-核糖单元转移到其底物上,包括 PARP1、PARP2、PARP5A、PARP5b;PARP13 是唯一一个在体外或体内都无法证明其催化活性的PARP家族成员(图1)[1]。
 图1. PARP家族
图1. PARP家族
monoPARP蛋白家族在与炎症性疾病、癌症和神经退行性疾病的发展相关的多种应激反应中发挥重要作用。
聚二磷酸腺苷(ADP)-核糖聚合酶7(PARP7)(也称为TiPARP)属于 monoPARP家族成员,催化ADP-核糖从烟酰胺腺嘌呤二核苷酸(NAD+)转移到蛋白质靶标上的氨基酸,这是一种称为单ADP-核糖基化(MARylation)。
PARP7最初被确定作为芳香烃受体(AHR)的合成激动剂(例如致癌物2,3,7,8-四氯二苯并对二恶英 [TCDD])强烈上调的基因。在某些细胞类型中(例如,小鼠胚胎成纤维细胞,MEFs),PARP7的表达为完全依赖于AHR[2]。
近期PARP7被报道为抑制IFN-I信号传导以响应胞质NA传感器配体和几种类型的RNA病毒的主要AHR调控基因。在MEFs中,在胞质NA传感器配体(即合成的 RIG-I 配体3pRNA和STING激动剂cGAMP)存在下敲除(KO) PARP7协同诱导IFN-表达[3]。
PARP7已被证明在肿瘤中过度活跃并在癌细胞存活中起关键作用,抑制PARP7可有效抑制癌细胞生长,恢复干扰素信号,有效防止癌细胞逃避免疫系统。
PARP7选择性抑制剂开发
开发PARP7或其它PARPs选择性抑制剂的主要障碍,都是PARP催化中的高结构保守域;虽然序列同源性只有大约50%,但它们的结构是高度保守的,特别是在NAD+结合位点(大多数PARP抑制剂的主要结合位点)。
到目前为止,只有一个PARP7选择性抑制剂RBN-2397(图2)进入临床I期研究,对PARP7的IC50小于3 nM, 较其他家族成员的选择性大于50倍以上。目前正在对RBN-2397开展单独(NCT04053673)或联合PD-1抑制剂帕博利珠单抗(NCT05127590)的临床研究。
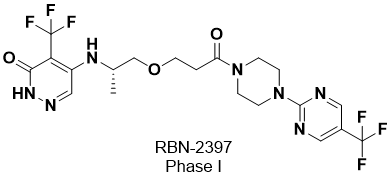
图2. RBN-2397的结构
RBN-2397由Ribon Therapeutics开发,通过HTS发现苗头化合物,针对烟酰胺腺嘌呤二核苷酸(NAD+)结合口袋关键区域的靶标相互作用进行了先导化合物的优化而得到。
PARP7可响应细胞应激(包括癌症中的基因组不稳定性)而被上调,并通过负向调节I型干扰素反应而抑制细胞应激反应(图3)。RBN-2397能够通过抑制肿瘤细胞中的PARP7,直接抑制细胞增殖并恢复I型干扰素信号,以刺激先天性或适应性抗肿瘤免疫应答。
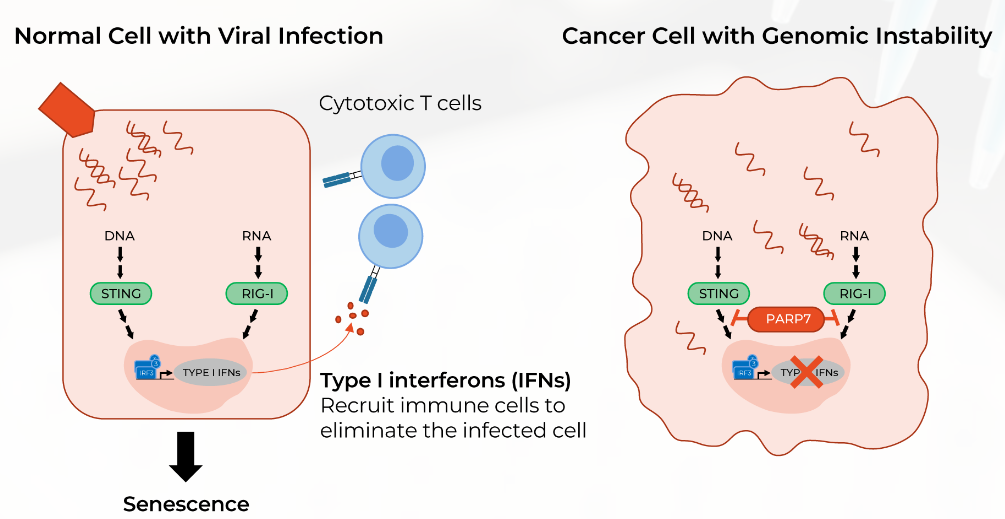
图3. PARP7负向调节I型干扰素反应
2021年2月2日,Ribon Therapeutics宣布已与Ono Pharmaceutical Co. Ltd.签订了独家许可协议,后者获得开发RBN-2397在日本,韩国,台湾和东盟国家(“小野地区”)商业化权益。
RBN-2397的PK性质不佳(图4),所以目前很多公司包括Ribon Therapeutics对RBN-2397进行me too,me better改造。
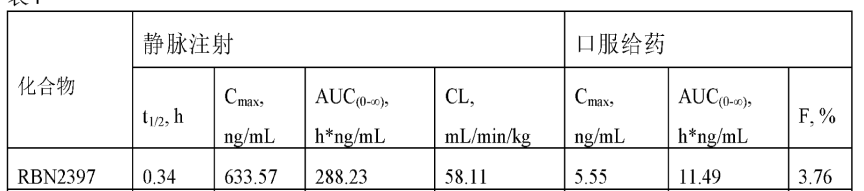
图4. RBN-2397在小鼠体内PK性质
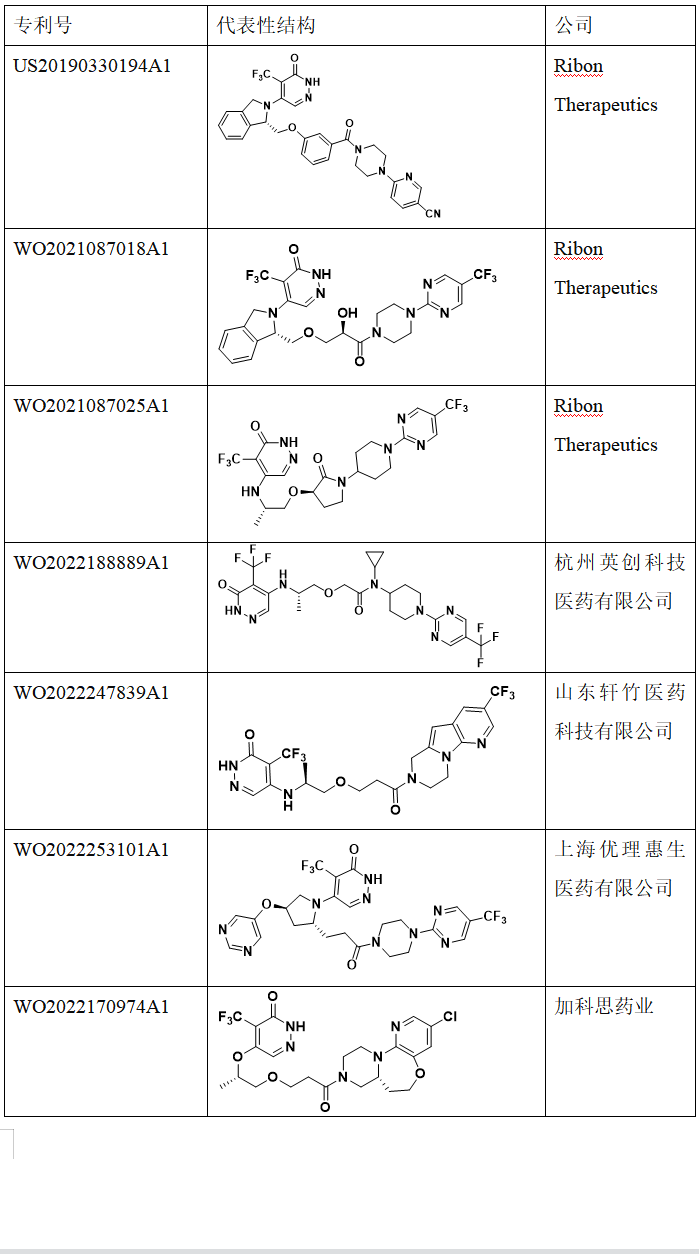
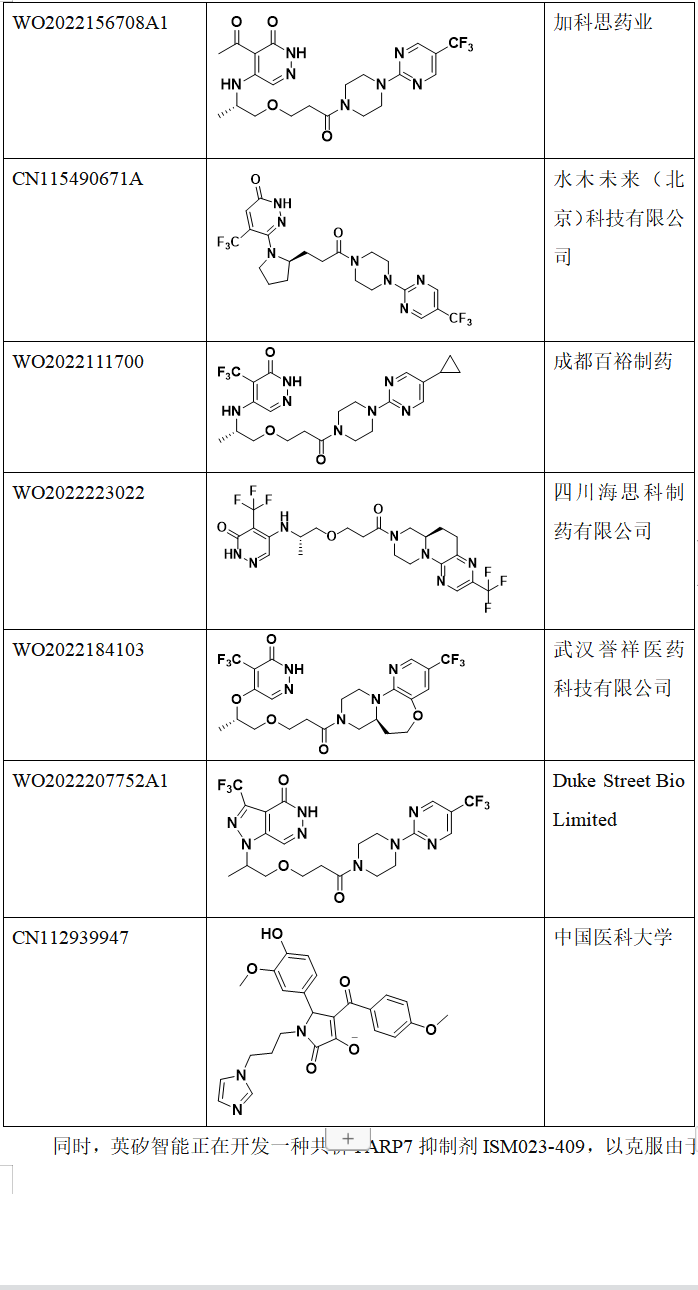
Ribon Therapeutics公司对PARP7抑制剂申请了多项专利,基本是围绕RBN-2397结构进行开发设计,如专利US20190330194A1,WO2021087018A1,WO2021087025A1等(表1)。除了Ribon Therapeutics之外,也有一些其它公司对RBN-2397进行me too,me better设计开发,如杭州英创科技医药有限公司,山东轩竹医药科技有限公司,上海优理惠生医药有限公司,加科思药业, 水木未来(北京)科技有限公司,成都百裕制药,四川海思科制药有限公司,武汉誉祥医药科技有限公司,Duke Street Bio Limited等(表1)。
除了以上公司,中国医科大学利用分子对接得到一些PARP7的优势构象,并测试了它们对不同肿瘤细胞的抑制活性,研究发现这些优势构象对肿瘤细胞都有很明显的抑制活性,可以用这些优势构象进行后续结构优化而得到与RBN-2397不同结构类型的PARP7抑制剂。
表1. PARP7抑制剂相关的专利
同时,英矽智能正在开发一种共价PARP7抑制剂ISM023-409,以克服由于高清除率而导致的潜在低暴露问题(图5)。

图5. ISM023-409对PARP7的分子和细胞活性
除了专利之外,近期也有一些PARP7选择性抑制剂相关的文献被报道,如发展在chemical cell biology和JMC上的选择性PARP7抑制剂。
Kirby等人之前在MARylating PARP中发现了一个独特的疏水腔,该腔毗邻NAD+结合位点的烟酰胺子口袋,可以被利用用于开发MARylating PARP的选择性抑制剂。
ITK6与PARP14(PDB:6FZM)的晶体结构显示Leu1782(人类PARP14)充当守门人,允许从抑制剂骨架连接疏水取代基进入疏水腔。
Sanderson等人认为设计策略的关键是确定包含位置的NAD+竞争性抑制剂骨架可以很容易地用取代基衍生化,该取代基将与PARP7中独特的疏水腔有利地相互作用[3]。
据报道,Rodriguez等人发现基于邻苯二甲嗪酮骨架的NAD+竞争性抑制剂Phthal01[4](图6A)是一种有效但非特异性的PARP7抑制剂。
PARP活性筛选和抑制剂测试测定(PASTA)结果显示Phthal01对三种 PARP(PARP1、PARP2 和 PARP7)表现出两位数的纳摩尔效力,但与所有其他 PARP 家族成员相比,对PARP7的效力至少高12倍。
为了将Phthal01转化为更具选择性的抑制剂PARP7,Sanderson等人设计并合成了Phthal01类似物KMR-206,在邻苯二甲嗪酮骨架的C-6位置引入丙炔基。
他们认为C-6丙炔基会占据PARP7中的疏水腔,但会在PARylating PARP中与谷氨酸发生冲突。他们使用PASTA来分析KMR-206以及最近描述的PARP7抑制剂RBN-2397对整个PARP家族的选择性(图6B)。
与Phthal01类似,KMR-206以中位抑制浓度有效抑制PARP7(IC50)的13.7 nM(图6B),然而,与Pthal01不同,KMR-206 显示大约75 倍PARP7对PARP2的选择性,并且在高达3 mM浓度时也不抑制PARP1。
跟之前报道类似,RBN-2397有效抑制PARP2(IC50 = 30.3 nM),但不能抑制大多数其他PARP家族成员。
接下来,他们试图确定KMR-206是否抑制细胞中PARP7的催化活性。他们发现用KMR-206处理表达GFP-PARP7的HEK 293T细胞导致 PARP7的MARylation的剂量依赖性降低(EC50 = 8 nM)(图6D和6E)。

图6. 强效膜渗透性PARP7抑制剂KMR-206的合理设计。A. Phthal01, KMR-206, and RBN-2397的结构,B. 全家族范围的 PARP 抑制剂筛选试验显示 KMR-206 对 PARP7 具有选择性,C. 与PARP7结合的KMR-206(橙色)诱导拟合同源模型显示丙炔基占据了由Ile631和D环中的疏水氨基酸(蓝色), D. KMR-206在HEK 293T细胞中以剂量依赖性方式抑制GFP-PARP7自体化,E. 使用 Bio-Rad 图像实验室软件对(D)的重复进行定量
中国药科大学Gu等人近期在JMC上报道了一种新型的PARP7抑制剂I-1,它具有高抑制能力(IC50 = 7.6 nM),对PARP7的选择性超过其他PARPs[5]。特别是,在CT26同基因小鼠模型中,I-1具有优异的药代动力学特性和低毒性,在不添加1-氨基苯并三唑(细胞色素P450的一种非选择性和不可逆抑制剂)的情况下,I-1在小鼠体内抗肿瘤活性(TGI: 67%)明显强于RBN-2397(TGI: 30%)。
他们的研究结果表明,I-1在肿瘤微环境中主要通过抑制PARP7发挥免疫激活剂的作用,这凸显了I-1作为肿瘤免疫治疗剂的潜在优势。
图7. I-1的结构改造和与PARP7对接的晶体结构
— 总结 —
PARP作为合成致死领域的热点一直受到广泛的关注,然而目前也出现对市场上已有的抑制剂的耐药性,未来对PARP领域的进一步开发就是针对各个亚型设计合成新型选择性抑制剂,希望提高选择性,减少毒副作用,开发更多新型结构的抑制剂,解决耐药性问题。
目前对PARP1,PARP7和PARP14研究较多,PARP7目前只有一款抑制剂进入临床研究,这一领域具有很大的研究价值和空间,国内很多药企也在积极布局,未来谁能胜出,敬请期待!
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言