
腓骨肌萎缩症(CMT):临床分型与诊断|疑难探究
2023-07-03 神经科学论坛 神经科学论坛 发表于上海

CMT最常见的遗传性神经疾病,通常出现在儿童时期,主要表现为肢体远端肌肉进行性无力和萎缩,伴有轻到中度感觉减退,腱反射减弱和足部畸形等。
论坛导读:Charcot-Marie-Tooth病(CMT)是最常见的遗传性神经疾病,通常出现在儿童时期,主要表现为肢体远端肌肉进行性无力和萎缩,伴有轻到中度感觉减退,腱反射减弱和足部畸形等。遗传性周围神经病是一组疾病,包括遗传性运动和感觉神经病(HMSN)、遗传性运动神经病(HMN)和遗传性感觉神经病(HSN)或遗传性感觉和自主神经病(HSAN)。其中,HMSN是最常见的类型,也被称为腓骨肌萎缩症(CMT)。
根据临床表现和电生理特征,CMT主要包括CMT1型(脱髓鞘型),神经传导速度(NCV)减慢(正中神经传导速度<38m/s),CMT2型(轴突型),神经传导速度正常或轻度减慢(正中神经传导速度>38m/s)。CMT多数呈常染色体显性遗传,也可呈常染色体隐性或X-连锁遗传。
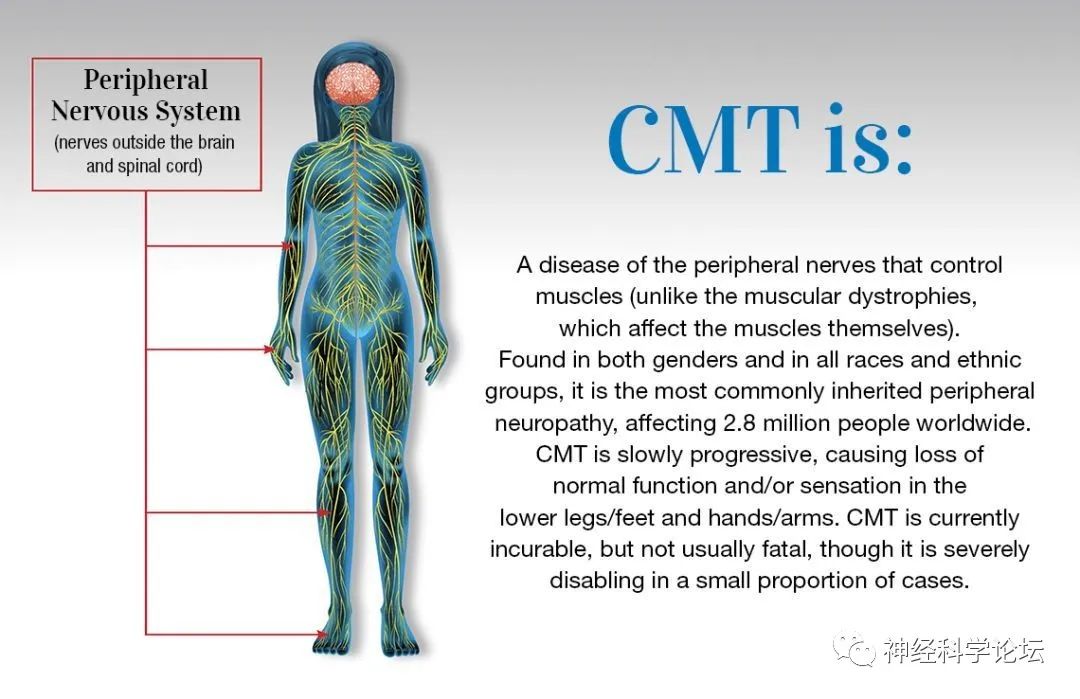
腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)是一组主要影响周围神经系统的遗传和表型异质性疾病。1886年,法国的Jean Marie Charcot和Pierre Marie以及英国的Howard Henry Tooth首先描述了这一疾病。CMT在临床、电生理、遗传和病理特征上是不同的。
然而,这些患者中的大多数在第一或第二个十年中表现为隐匿性发作和缓慢进行性虚弱,开始于下肢,后来累及上肢。CMT是人类最常见的遗传性神经病,分为轴突型、脱髓鞘型或髓鞘形成障碍型,或中间型。CMT主要是一种周围神经系统疾病,患者通常表现为脱髓鞘性神经病或轴突神经病或两者兼有的特征。很少有患者表现出中枢神经系统受累的特征。帕金森综合征、失语症和家族性癫痫综合征以前曾作为与CMT4J型J相关的病例报告出现。目前临床主要有5个分型:
- 脱髓鞘型(CMT 1):在10岁以内发病慢性进展性病程;周围神经对称性肢体远端肌无力和肌萎缩自足和下肢开始;常伴脊柱侧弯垂足呈跨阈步态;部分病人仅有弓形足或神经传导速度减慢甚至无临床症状。运动和感觉神经传导速度(nerve conduction velocity,NCV)明显下降,NCV<38m/s(正常>40,~45m/s) ;
- 轴索变性型(CMT 2): 发病晚成年开始出现肌萎缩症状及出现部位与CMT1型相似程度较轻;运动NCV正常或接近正常CSF蛋白正常或轻度增高;神经活检主要为轴突变性。髓鞘相对保留,NCV正常或接近正常;
- CMT 3 :婴儿期起病的严重脱髓鞘性CMT ,即Dejerine-Sottas病(DSD);
- CMT 4 :大部分隐性遗传性CMT ;
- CMTX :X连锁遗传 。
CMT是最常见的遗传性周围神经病,患病率为1/2500。疾病发作可发生在从婴儿期到成年晚期的不同生命阶段,但症状大多出现在5至25岁之间。一项纳入的31项meta研究中,21项研究了全年龄人群和各种类型的CMT综合征,其他研究报告了特定的疾病亚型或成人或非成人人群。全年龄人群的合并患病率为17.69/100,000 ,CMT1的患病率显著高于其他亚型(P < 0.001)。年龄≥ 16或18岁的人群的患病率高于<16岁的人群,男性高于女性(17.95/100,000),北欧高于其他地区。
CMT大多数患者表现为高足弓(pes cavus畸形)、踝关节背屈无力、深部腱反射减弱或缺失、远端肢体肌肉无力和萎缩以及轻度至中度远端感觉丧失。某些CMT亚型还可能伴有听力损失、视神经病变或其他中枢神经系统功能障碍。CMT进展缓慢,残疾随着时间的推移逐渐积累。根据遗传模式(常染色体显性、常染色体隐性和X-连锁)、运动神经传导速度和原发性缺陷类型(主要是脱髓鞘或髓鞘形成障碍,主要是轴突或中间类型)对CMT进行分类,CMT1和CMT4重组了主要的脱髓鞘和髓鞘形成障碍形式,CMT2重组主要的轴突形式,I-CMT重组了中间形式。此外,CMT的遗传多样性导致了几种具有独特表型特征的亚型。
周围神经系统的髓鞘神经胶质施万细胞多次包裹轴突,以构建其髓鞘。髓磷脂对于轴突完整性和快速轴突电位传播至关重要。然而,在包括脱髓鞘和髓鞘形成障碍的CMT在内的几种神经病中,髓鞘缺乏或功能障碍。CMT的脱髓鞘或髓鞘形成障碍形式构成大多数疾病病例,最常见的是由于以下三种髓鞘基因的突变:外周髓鞘蛋白22、髓鞘蛋白零和缝隙连接β1(编码连接蛋白32 ),分别导致CMT1A型、CMT1B型和X连锁CMT1型。由此引起的髓鞘结构和功能的紊乱导致轴突脱髓鞘或髓鞘形成障碍,并导致受影响患者的严重残疾。
很多类型的CMT电生理特点常介于脱髓鞘型和轴索损伤型之间,NCV (35~45m/s)更细致的分型常由致病基因及其位点决定 。目前超过120个致病基因已被确定,其中许多基因有几种变异。因此,不同类型CMT的病理机制是不同的。它们可以影响致密和非致密髓鞘、细胞信号、转录因子、线粒体、细胞骨架、转运蛋白、通道、蛋白质质量控制机制以及轴突运输。然而,90%的CMT患者患有脱髓鞘或髓鞘形成障碍形式的CMT,其特征在于上肢运动神经传导速度低于35米/秒,最常见的形式是CMT型1A (CMT1A)、CMT型1B (CMT1B)和X-连锁的CMT 1型(CMT1X)。在这些脱髓鞘或髓鞘形成障碍的CMT过程中,许旺细胞(SCs)不会完全分化、去分化或产生不稳定的髓鞘。所以髓鞘要么不形成,要么退化,轴突脱髓鞘,最后退化。一项在31例CMT病例研究中,基因突变检出率为42%,最常见的遗传变异是PMP22重复。家系分析显示在NEFL和PMP22基因中分别有两个新突变c.64C > A (p.P22T)和c.281delG (p.G94Afs*17)。
CMT1A是一种脱髓鞘和髓鞘形成障碍,CMT1A占所有CMT1的70~80%。最近从CMT1A患者的电生理数据中发现,CMT1A可能主要是髓鞘形成障碍而不是脱髓鞘,洋葱球形成将是发育期间轴突周围异常SC组织的结果。
CMT1B占CMT脱髓鞘或髓鞘形成障碍的10%,是由影响编码P0的MPZ基因的突变引起的。CMT1B通常根据发病年龄、临床和病理特征分为两类。早发型的特征在于儿童期的严重脱髓鞘和残疾,以及成年期疾病进展的减缓。相比之下,成人形式主要是轴突神经病,其特征在于快速进展但伴有非常轻微的脱髓鞘。
CMT1X是一种X连锁的显性CMT,占所有CMT病例的5-10%。由编码Cx32的GJB1基因突变引起。男性比女性受影响更严重,疾病在儿童时期就已经开始发病。与其他形式的脱髓鞘CMT不同,CMT1X被认为是一种混合的轴突和脱髓鞘病变,轴突缺失在儿童期就可能发生。患者在代谢应激条件下也可出现脑病综合征。
鉴别诊断首先需要与Krabbe 脑白质营养不良、异染性脑白质营养不良、线粒体病、遗传性痉挛性截瘫和遗传性共济失调等遗传性周围神经病变鉴别。它们除具有周围神经病以外,还有神经系统其他部位和非神经组织器官受累的表现,而CMT 较少有周围神经以外的其他系统受累。另一些以周围神经受累为主的遗传性病,如远端遗传性运动神经病(dHMN)、Refsum病、家族性淀粉样变性、巨轴索神经病和遗传性压迫易感周围神经病等,需要在临床和电生理检查基础上,选择必要的生化检验、神经活检病理和基因检测来加以鉴别。此外,还需要与远端型肌病和下运动神经元综合征(如脊肌萎缩症)相鉴别,肌电图及必要的肌肉活检病理及基因检测有助于鉴别诊断。
其次,CMT 需要与获得性周围神经病相鉴别,如CIDP(慢性炎性脱髓鞘性多发神经根周围神经病)、副蛋白血症相关周围神经病,轴索性如中毒、代谢相关周围神经病和多灶运动神经病等。CMT通常在青少年或幼年起病,起病年龄晚需警惕获得性周围神经病。CMT 多起病隐匿、数年内缓慢加重,而获得性周围神经病多病程较短。查体发现弓形足、锤状趾、鹤腿症状提示CMT 可能性大。脱髓鞘型CMT 的运动神经传导速度通常均匀减慢,若出现明显的波形离散、传导阻滞,通常提示CIDP 可能性大。CMT 的脑脊液蛋白可轻度升高,但若明显升高
(如>1g/L)则需考虑CIDP 等获得性周围神经病可能。
目前临床上没有治愈或减缓疾病进展的治疗方法,但是,科学发现导致对疾病的病理机制和潜在治疗策略的更好理解。有研究发现中草药、免疫抑制剂、维生素及物理康复方法等综合治疗可延长病程,减轻残疾发生率,对症和支持疗法有助于减缓症状,如足下垂或足畸形穿矫形鞋, 严重的足畸形引起疼痛选择外科手术,避免应用可能导致神经损害的药物,如长春新碱等。由于CMT病程进展缓慢大多数患者可存活数十年,对症治疗有助于提高患者生活质量。
参考文献
Hertzog N, Jacob C. Mechanisms and treatment strategies of demyelinating and dysmyelinating Charcot-Marie-Tooth disease. Neural Regen Res. 2023 Sep;18(9):1931-1939.
Nguyen-Le TH, Do MD, Le LHG, Nhat QNN, Hoang NTT, Van Le T, Mai TP. Genotype-phenotype characteristics of Vietnamese patients diagnosed with Charcot-Marie-Tooth disease. Brain Behav. 2022 Sep;12(9):e2744.
Ma M, Li Y, Dai S, Chu M, Sun L, Liu L, Zhou JC. A meta-analysis on the prevalence of Charcot-Marie-Tooth disease and related inherited peripheral neuropathies. J Neurol. 2023 May;270(5):2468-2482.
Chaudhuri J, Dutta AK, Biswas T, Biswas A, Ray BK, Ganguly G. Charcot-Marie-Tooth disease type 4J with spastic quadriplegia, epilepsy and global developmental delay: a tale of three siblings. Int J Neurosci. 2022 Aug;132(8):783-786.
Morena J, Gupta A, Hoyle JC. Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci. 2019 Jul 12;20(14):3419.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言








#腓骨肌萎缩症##疑难病例诊断#
39