炎症在阿尔茨海默病发病机制中担当了什么样的角色?
2023-02-18 ANDs ANDs 发表于安徽省

阿尔茨海默病:是一种进行性发展的致死性神经变性性疾病,1906年由AlosisAlzheimer首先描述。临床上以进行性智能减退、行为紊乱和认知功能障碍为主要特征。
阿尔茨海默病 ( Alzheimer’s disease, AD) 是一种进行性发展的致死性神经变性性疾病,1906年由AlosisAlzheimer首先描述。临床上以进行性智能减退、行为紊乱和认知功能障碍为主要特征。导致日常生活能力进行性减退,并有各种神经精神症状和行为障碍。据中国阿尔茨海默病协会2011年的公布调查结果显示,全球有约3650万人患有痴呆症,每七秒就有一个人患上此病,平均生存期只有5.9年,是威胁老人健康的“四大杀手”之一。
大量研究资料表明炎症在AD的发病机制中的核心地位。尤其脑内的慢性炎症不仅是beta-amyloid peptide (Ab ) 产生和沉积、老年斑 (senile plaques, SP ) 形成的主要病理生理机制,而且也是tau高度磷酸化、神经原纤维缠结 ( neurofibrillary tangles,NFTs ) 病变、神经元变性及乙酰胆碱含量显著减少的主要因素。然而,炎症双刃剑作用,在我们谈论炎症在AD病理生理作用机制时,往往忽略了其保护作用。所以,本文就目前国内外对炎症在AD发病机制中的作用进行综合,结合炎症的保护作用和AD抗炎临床研究证据,分析和展望炎症在AD发病机制中的作用进行综合,结合炎症的保护作用和AD抗炎临床研究证据,分析和展望炎症在AD发病机制中的角色。
 炎症在AD发病机制中的核心地位证据
炎症在AD发病机制中的核心地位证据
炎症是机体对外来刺激产生的一种病理反应过程,症状表现为局部的红肿热痛,病理检查可发现有大量炎症细胞如粒细胞,巨噬细胞的局部浸润和组织坏死。在这一过程中,一些炎症细胞因子主要包括白细胞介素( interleukin, IL )、肿瘤坏死因子 ( tumornecrosis factor, TNF )、干扰素 ( interferon, IFN)、集落刺激因子( colony stimulating factor, CSF )、趋化因子(chemokine )、淋巴细胞产生的淋巴因子、单核巨噬细胞产生的单核因子等,它们在免疫系统中起着非常重要的调控作用,如IL-1、IL-6、TNF-α等可促进炎症细胞的聚集、活化和炎症介质的释放,加重炎症症状。在AD中都可检测到上述细胞因子的水平升高。用某些细胞因子给AD动物模型注射,可直接诱导炎症现象,这些实验充分证明细胞因子在AD炎症过程的重要促进作用。
炎症和Ab沉积的相互作用
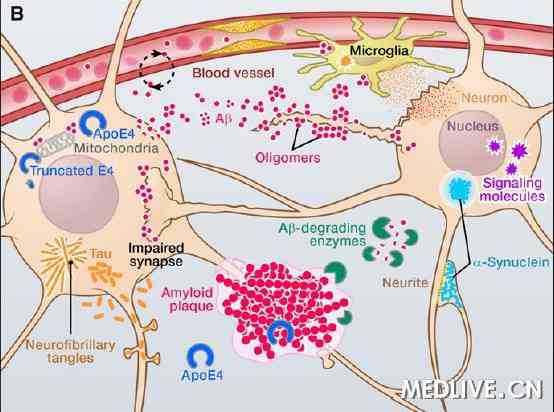
炎性反应是Ab产生和沉积的核心病理机制,主要表现为白介素-1 ( IL-1 )、白介素-6 ( IL-6) 和肿瘤坏死因子-α ( TNF-α ) 等炎性因子在AD中促进了Ab的产生和SP的形成[1-3]。
IL-1包括IL-1α和IL-1β,前者主要位于细胞内或表达于细胞表面,发挥分泌信使功能,后者则释放到细胞外,通过作用于其他细胞而发挥作用。在AD中,IL-1β的过度表达促进了Ab的沉积和SP的形成。研究也表明IL-1β的过度表达可能发生于SP早期,可通过蛋白激酶C( protein kinase C,PKC )、 cdk5/p25、GSK-3β、p38-MAPK、NF-kB等途径促进APP的合成和分泌裂解[2, 4],从而进一步促进Aβ的产生和沉积,因此IL-1的过度表达在SP早期形成过程中起决定作用。
在中枢神经系统,IL-6主要由神经元和胶质细胞合成,IL-6的生物学活性是通过IL-6受体介导的。基础研究证明IL-6不仅可以促进神经元上调表达β-APP[5],同时也可以上调β-分泌酶的表达(BACE)[6],导致Aβ生成与沉积异常增加,从而诱发AD[7]。
TNF-α是主要由单核细胞和巨噬细胞(星形胶质细胞、小胶质细胞等)产生的细胞因子,具有IL-1和其他细胞因子的特性,主要作用是调节免疫细胞的功能[8]。越来越多的研究表明,TNF-α在Aβ生成过程中起着重要的推动作用[9-13]。小胶质细胞TNF-α的过度表达可以导致类似于AD的Aβ积聚,而且也有资料表明Aβ的产生依赖于肿瘤坏死因子受体信号通路[14]。
Aβ沉积引起的脑内慢性炎症反应可能是AD发病的核心病理机制之一。一方面Aβ本身担当了炎症的直接触发剂大的角色,另一方面Aβ沉积使得神经胶质细胞活化。在AD炎症过程中,小胶质细胞则是其最主要的炎症细胞,小胶质细胞被Aβ激活,释放大量致炎性细胞因子和神经元毒性介质,从而诱发脑内炎症反应,导致神经元损伤、死亡。Aβ的持续存在,小胶质细胞被持续激活释放炎性细胞因子,导致炎症持续发生的恶性循环,最后导致AD的发生和发展。AD患者脑内细胞因子水平持续增高,主要由于激活状态的小胶质细胞产生。
炎症是NFTs形成的催化剂
NFTs和SP是AD的两大病理特征。NFTs在神经元细胞体以及轴突和树突内形成神经原纤维包含体,神经原纤维包含体内的基本成分是双螺旋状或长15nm 的直的神经原纤维。这些神经原纤维由一大类蛋白质构成,包括微管蛋白、tau 蛋白、泛素、中间细丝蛋白和聚糖类,其中最主要的成分是磷酸化的不溶性tau 蛋白。正常情况下,tau蛋白位于轴索和神经元胞体中,多与细胞内微管上的微管蛋白相结合,呈可溶性,有促进微管的聚合和稳定的作用。而AD 时,tau蛋白呈现过度磷酸化,从微管上解离,由可溶性的tau 蛋白变为不溶性的tau蛋白,进而形成双螺旋状或直的神经原纤维,导致神经原纤维缠结。
大量资料已经证实炎症因子可以促使tau 呈现过度磷酸化,AD脑内炎症因子表达水平与tau蛋白病变有着密切的相关性。给3xTg-AD 转基因鼠腹腔注射脂多糖( LPS ) 诱发神经炎症,APP 未受影响,但是tau蛋白呈现过度磷酸化,而且进一步证实tau蛋白过度磷酸化是由于p25 片断明显增高诱发cdk5激活这一途径实现的[15, 16]。研究也表明炎症诱导的tau过度磷酸化可以通过激活GSK-3β这一机理实现[17]。同时,tau过度磷酸化和NFTs也是AD脑内炎症的引发促进者[18]。
炎症是AD神经元变性的杀手
胆碱能系统功能缺陷与AD密切相关,AD脑内隔区、Meynert基底核等部位的胆碱能神经元变性明显减少。以IL-1β和NTF-a为代表的炎性细胞因子参与许多神经病理过程,包括脑缺血引起急性神经变性、AD等的慢性神经变性。AD脑内慢性炎症首先通过氧化应激途径成为胆碱能神经元变性的杀手,其次加速了正常神经元的凋亡,促进了AD脑内神经元的大量丢失。同时AD 脑内炎症和神经变性使得游离于神经元之外的AChE ( acetylcholinesterase, AChE ) 活性相对显著提高,Ach大量水解,使皮层ACh活性进一步降低。ACh减少的程度与AD认知功能损害的严重性呈正相关,最后导致学习记忆能力缺陷的表现[19, 20]。
大量证据表明脑老化和神经变性伴随着神经细胞内钙稳态的失调和细胞钙调节能力降低而发生。细胞膜上钙通道开放和细胞内钙库近年来钙代谢及其体内平衡与老化及神经系统退变性疾病之间有着密切的联系。神经炎症不仅可以通过氧化应激参与了AD线粒体变性,也能够通过影响神经元内钙稳态的失调和细胞钙调节能力降低,参与了AD的神经变性过程。
炎症破坏突触可塑性是AD认知功能损害的直接推动者
突触可塑性是学习和记忆的基础,突触可塑性最直接的证据是突触传递长时程增强 ( long-termpotentiation,LTP ) 现象[21, 22]。LTP在神经功能可塑性方面起了重要的作用,是学习记忆的电生理基础。LTP被认为可直接反映突触水平信息贮存过程,学习记忆同海马神经元突触可塑性密切相关,LTP被用来作为评价海马神经元突触可塑性的重要指标。AD的早期临床表现是单纯的记忆功能损伤,认知障碍进行性加重,并出现神经退行性改变。突触功能障碍及缺失是发生于阿尔茨海默病AD早期的病理过程,是AD疾病过程中的重要神经病理改变之一,新皮质、海马的联合区的突触的完整性受损、可塑性异常、密度下降被认为是AD认知障碍的发病基础[23, 24]。研究发现持续的炎症可导致突触的可塑性下降,是LTP受损的直接推动者[25, 26]。分子生物学证据表明突触素和微管相关蛋白-2 ( microtube-associated proteins-2, MAP-2 ) 均是突触结构可塑性的重要指标,当发生脑内炎症的病理性损伤时,突触素和MAP-2的表达下降,是记忆认知功能损害的机制之一。炎症也可以下调记忆蛋白cAMP效应元件结合因子( cAMP response element binding, CREB ),影响LTP的功能维持和长时程记忆的形成[27, 28]。同时,在AD中Ab产生和沉积诱发的炎症是突触结构改变的促发者之一。突触的结构尤其是树突棘是突触可塑性的基础,大量研究发现神经炎症与神经元突触和树突棘结构的改变并存,且神经炎症发生后可观察到有突触和树突棘数量、密度的下降[29]。
炎症在AD中的脑保护作用证据
众所周知,炎症是指具有血管系统的活体组织对各种损伤因子的刺激所发生的一种以防御反应为主的基本病理过程,是机体对于刺激的一种防御反应。通常情况下,炎症是有益的,是人体的自动的防御反应。在AD脑内存在大量炎症特性,如炎性细胞因子及趋化因子增加、损伤部位活化的小胶质细胞积聚等。AD的神经炎症反应主要由不溶性Ab沉积而成,Ab激活小胶质细胞和星形胶质细胞然后引起神经元退行性病变有关。小胶质细胞可聚集在含SP的细胞外发挥吞噬功能以降低Ab的沉积,这种作用说明了神经炎症对AD的有利方面[30, 31]。一方面炎症反应可以去除组织细胞碎片、有害蛋白质和受损神经元释放的毒性物质,甚至通过分泌神经营养因子等以促进神经元的修复,这些作用均有利于受损神经元的存活及功能上的恢复。
许多研究报道指出炎性细胞因子的表达增多是神经保护性的,属防御反应范畴。也有很多的研究认为小胶质细胞活化和炎症反应对AD有利,研究证实小胶质细胞的活化对Ab具有吞噬作用。最近研究发现在APPswe/PS-1dE9 AD动物模型中,慢性IL-1β介导的神经炎症并没有诱导显著地神经变性,反而降低了Aβ的产生和减缓了SP的形成[32]。
因此,也有学者推测炎症细胞因子在AD脑中的生物学作用是双重的,即有防御抑制损伤的一面,又有促进神经损伤的一面。主要取决于炎性细胞因子表达量的多少,也取决于它们所诱导的继发性反应和反应产物的表达量的多少。
非甾体抗炎药物临床药物试验失败的反思
大量研究证实神经炎症在AD发病机制的核心地位是非甾体抗炎药物临床应用的理论依据。研究发现长期使用非甾体抗炎药可以降低AD的发病率,长期使用非甾体抗炎药( non-steroidalanti-inflammatory drugs, NSAIDs )的关节炎患者AD的发病率显著降低。而且也认为NSAIDs降低AD发病率的机理主要是NSAIDs抑制小胶质细胞活化和其炎症因子的表达。
但是最近几年临床研究结果却与实验研究的期望相反,有的学者甚至提出NSAIDs预防AD的机理可能与其抗炎作用无关,可能与NSAIDs (如舒林酸、消炎痛等)能够降低AD的主要病理因素Ab有关[33],也有报导长期NSAIDs并没有显示出对AD病理的显著改善[34]。阿司匹林是临床最常用的NSAIDs,虽然对防治心脑血管病的效果已经被公认,但对AD患者并不有利,不能改善和延缓AD认知功能损害程度,反而增加AD患者消化性溃疡发生和严重上消化道出血的风险[35, 36],增加了自发性脑出血的风险[37],故不主张推荐用于治疗AD患者。普通NSAIDs萘普生和COX-2抑制剂罗非昔布未能延缓AD的进程,反而加速了Ab的沉积和SP的形成[36, 38]。研究也证明大剂量服用NSAIDs的人发生痴呆的风险比那些小剂量或者是没有服用NSAIDs的人要高,而且大剂量服用NSAIDs的病人患痴呆的风险实际上更高。
NSAIDs在治疗AD临床药物试验上的失败也提醒人们思考炎症在AD发病过程的真实作用。机体器官功能的正常运行无时无刻都与炎症有关,炎症是病理过程也更是生理过程的必需。AD的炎症基础理论的提出多是建立在大量的基础实验上,大多数是外源性因子引发的急性实验,这与AD的慢性神经变性过程是不匹配的。NSAIDs在治疗AD临床药物试验上的失败也在一方面提示炎症也许不是AD的核心病理生理机制。
鉴于AD是和年龄相关性疾病,老化和年龄被认为是“程序机制”疾病,那么AD能否也可以被认为是与“程序机制”理论有关。慢性炎症理论似乎是老化和AD的共同通路,能否揭示慢性炎症和“程序机制”理论之间的联系成为AD研究的一个突破?
展望
现代生物医学关于AD的炎症机制相关研究及防治策略可谓面面俱到,正反有别,论据论证洋洋洒洒,各家自圆其说。关于防治NSAIDs在治疗AD患者过程中,一个个被运用,又一个个被否定。如何揭开炎症在AD发病机制中的真面目有待进一步思考。能够揭开炎症在AD发病机制中真实的作用机理,将会使得所有的有关AD炎症理论和治疗策略就会变得豁然清晰。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言