
肌萎缩侧索硬化(ALS):靶向基因治疗|困局与突破
2023-05-13 神经科学论坛 网络 发表于上海

目前ALS相关研究虽然已经步入临床治疗阶段为单基因突变患者提供个性化的ASOs治疗策略有可能成为ALS的常规治疗方案。
论坛导读:自从1993年发现肌萎缩侧索硬化(ALS)基因突变中的Cu/Zn超氧化物歧化酶(SOD1)第一个遗传异常以来,已经鉴定出50多个基因是ALS和ALS/额颞叶痴呆(FTD)谱系疾病的原因或修饰物。C9orf72、SOD1、TAR DNA结合蛋白43 (TARDBP)和融合肉瘤(FUS)基因的突变是四种最常见的突变。在过去的三十年里,世界范围内已经做出了巨大的努力来揭示ALS/FTD中这些基因突变的发病机制背后的生物学途径。因此,已经广泛研究了靶向病因基因(即基因疗法)以抑制它们的毒性作用。2023年4月25日,Biogen/lonis联合宣布FDA已同意加速批准用于治疗超氧化物歧化酶1(SOD1)突变所致肌萎缩侧索硬化(ALS)的反义寡核苷酸疗法Tofersen上市,这是首款针对ALS的基因靶向疗法。

王共强 主任医师 硕士生导师
安徽中医药大学神经病学研究所
肌萎缩侧索硬化(Amyotrophic lateral sclerosis,ALS)是一种由运动神经元变性引起的毁灭性疾病,大多数患者在症状出现的2-3年内死于神经肌肉呼吸衰竭。ALS在全球范围内发生,发病率每年约为2/100,000人,患病率为6~9/100,000和大约1/350的终生风险。正如所有主要的神经退行性疾病一样,由于多种原因,疾病缓解疗法的发展已被证明具有挑战性。然而,ALS是少数被批准进行疾病缓解治疗的神经退行性疾病之一。在过去的10-15年里,在ALS临床前模型、遗传学、病理学、生物标记、成像和临床读数方面取得了重大发现和进展。与此同时,新的治疗模式正被应用于高度未满足的医疗需求领域,包括神经退行性疾病。
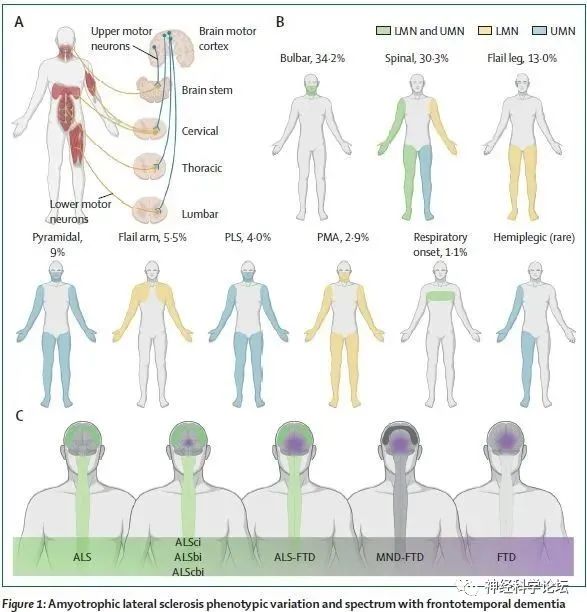
肌萎缩侧索硬化(ALS)和额颞叶痴呆(FTD)是两种致命的神经退行性疾病,属于一个疾病谱,具有共同的临床、遗传和病理学发现,大约5%的ALS患者出现明显的FTD特征,但详细的神经心理学评估显示,多达50%的患者出现更为微妙的额叶和颞叶功能障碍。ALS影响运动皮层中的上运动神经元(UMNs)和脑干和脊髓中的下运动神经元(LMNs)。特征性临床表现包括扩散至所有四肢和延髓肌肉的局部无力和反射亢进。疾病范围从主要的UMN病(原发性侧索硬化症[PLS])到主要的LMN病(进行性肌萎缩[PMA])不等。大约50%的ALS患者可能出现不同程度的认知功能障碍,大约15%的FTD患者可能出现ALS表型。超过97%的ALS患者和大约50%的FTD患者在受影响的神经元和神经胶质细胞中都有焦油DNA结合蛋白43 (TDP-43)聚集的组织病理学发现。尸检结果还显示,皮质脊髓束和脊髓/延髓运动神经元的变性伴随着中枢神经系统(CNS)内免疫细胞(即小胶质细胞、星形胶质细胞和少突胶质细胞)的激活。ALS表现为多种表型,延髓症状和脊髓型(颈椎和腰椎)ALS是最常见的表现,各占病例的四分之一。
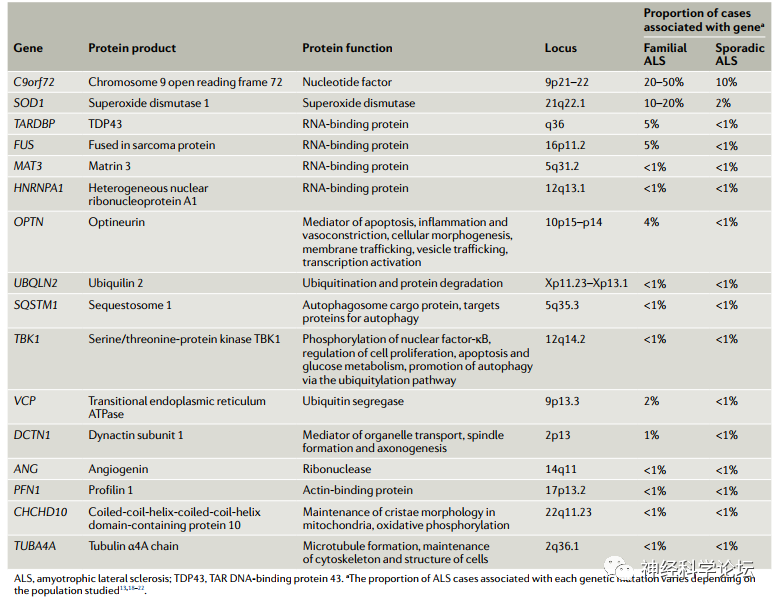
虽然绝大多数ALS病例是散发性的(sALS),但约10%的病例是家族性的(fALS) ,主要为常染色体显性,很少为X连锁或隐性遗传。1993年,胞浆铜/锌超氧化物歧化酶(SOD1)基因突变被确定为ALS中的第一个遗传异常。从那时起,为确定ALS病理学中涉及的突变基因所做的巨大努力已经确定了超过50种基因和120种遗传变异,它们增加了ALS的风险或改变了ALS的表型。对这些突变ALS基因潜在的分子途径的分析大大提高了我们对fALS和sALS发病机制的认识,从而为潜在的治疗靶点提供了新的见解。目前研究在家族性ALS中SOD1占10~20%、 C9orf72占20~50%、TAR DNA结合蛋白43 (TARDBP或TDP-43)占5%和肉瘤融合(FUS) 占5%基因的突变被发现是四种最常见的突变,涉及70%以上的家族性ALS病例。
但是,ALS遗传学是高度动态的,每年通过使用不断增长的数据集和尖端方法,一系列新的ALS相关基因和下游致病机制不断被发现。最近的疾病基因发现构成了候选和风险基因SPTLC1,KANK1,CAV1,HTT和WDR7,以及7个新的风险基因位点。细胞类型和功能富集分析揭示了ALS中选择性运动神经元脆弱性的遗传基础,证明了ALS相关基因在皮质运动神经元中的高表达,并强调了细胞自主过程的致病意义。关于已知ALS基因/蛋白,特别是C9orf72、TDP43、ANXA11和KIF5A,已经获得了主要的病理机制见解。第一个基于ASO的家族性als基因特异性治疗试验产生了不明确的结果,强调了与SOD1和C9orf72突变相关的病理机制的重新评估。
因此,开发转基因动物模型和靶向异常基因,即基因治疗已经在世界范围内进行了研究,以便将这些实验性基因治疗转化为临床设置。虽然对不同物种的临床前研究可能具有挑战性,因为它们可能无法真正代表确切的人类表型,但这些研究的结果令人鼓舞,并导致在ALS临床试验中启动了其中一些策略,包括四个主要策略:
-
使用微小RNA或反义寡核苷酸(ASOs)去除或抑制异常转录的RNA; -
使用RNA干扰(RNAi)降解异常mRNA; -
减少或抑制突变蛋白(例如,使用针对错误折叠蛋白的抗体); -
使用诸如聚集的规则间隔短回文重复序列(CRISPR)/CRISPR相关蛋白(CRISPR/Cas)的方法编辑DNA基因组。
这些研究的有希望的结果已经导致了这些策略中的一些在ALS临床试验中的应用,特别是对于C9orf72和SOD1。
对于绝大多数遗传疾病,甚至是单基因疾病,仍然缺乏明确的治疗方法。一般来说,需要几年的研究才能了解致病基因的正常功能及其发病机理的分子途径。即使有了这些知识,开发针对异常基因的技术,尤其是那些具有显性特征的基因,可能需要更长的时间。ALS也是如此,其中10-15%的病例是显性的高外显率基因变异。总的来说,目前有四种方法可以抑制致病基因的毒性作用:
-
微小RNA或反义寡核苷酸(ASOs设计成与靶序列配对并激活RNA降解的互补DNA或RNA序列)用于切除从基因转录的RNA:施用ASOs,其是靶向/改变mrna的合成核酸,在治疗儿童的其他神经肌肉疾病中显示出有希望的结果,例如脊髓性肌萎缩症(SMA)和杜氏肌营养不良症(DMD)。这完全改变了最初的疾病轨迹,促使FDA批准了两种aso,nusinersin (Spinraza)和eteplirsen (Exondys51),分别用于治疗1型和2型SMA和DMD的一个子集;然而,这些尚未在成人型SMA 3和4中进行测试,它们在成人疾病中的效用尚不清楚。总体而言,aso要么通过募集核酸内切酶RNase H选择性降解mrna,要么阻止RNA与RNA结合蛋白(RBP)的相互作用,从而调节它们的剪接/加工而不降解。
-
过量突变蛋白的减少(例如,免疫介导的减少)。
-
使用小分子干扰转录过程。
-
体细胞诱变,基因回复到野生型的反向突变。
目前的几份研究报告已经证明了这些方法中的前三种是可行的,最后一种方法的最大优点是突变DNA的校正消除了下游的异常,并且至少在理论上是一次性的干预。
反义寡核苷酸药物(ASO)是小分子,通过靶向RNase H1依赖性降解途径来介导细胞质mRNA和核保留RNA的降解,进而减少细胞蛋白质合成。在对其他神经退行性疾病(如SMA)的研究中,ASO已经显示出降低全因死亡率的有效性。在ALS靶向SOD1中使用ASO的第一次试验中,研究人员对大鼠和恒河猴进行了ASO鞘内注射,证明了CNS的全面覆盖,并发现ALS大鼠模型中疾病进展的减缓。在人体试验中,鞘内注射靶向SOD1的ASOs的I期试验(ISIS 333611在11.5小时内输注不同剂量的0.15、0.50、1.50和3.00mg;(Clinicaltrials.gov识别号:NCT01041222) 被认为是安全且耐受性良好的。
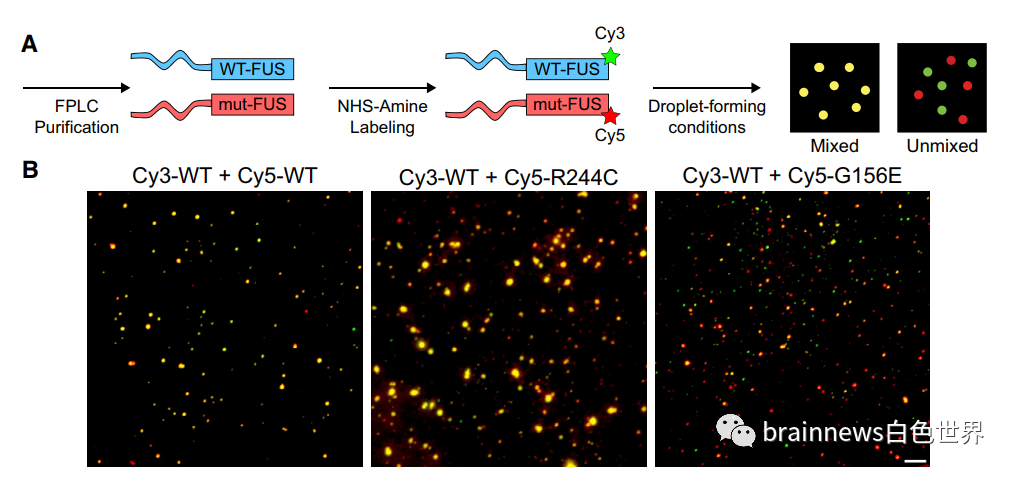
ASO通过阻止细胞产生异常蛋白质来抑制肉瘤融合蛋白(FUS)基因的致病作用,从而延缓了小鼠和FUS突变ALS患者的运动神经元退化速度。FUS基因具有重要的生物学功能。先前的研究证明,FUS基因突变会导致异常蛋白质的产生并聚集成团,从而引起运动神经元退化及死亡。而ASO药物通过靶向抑制有毒FUS蛋白形成的方式进行基因治疗,从而起到延缓以及防止疾病进展的治疗作用。针对FUS基因突变的ASO药物治疗,在未来确实有可能成为延缓ALS患者疾病进展的一个有效药物。
第二代ASO完成了I/II期试验(ClinicalTrials.gov识别号:NCT02623699),并发现了一种剂量依赖性疗效,最高剂量为100 mg,显示出在降低CSF SOD1浓度方面的最大效果,特别是在疾病快速进展中。然而,由于最初试验的参与者人数较少,BIIB067被延长至3期临床试验;然而,在28周的治疗中测量ALS快速进展者的疾病进展的主要结果没有达到统计学显著性。一项随访7年的BIIB067长期3期临床试验目前正处于规划阶段(Clinicaltrials.gov识别号:NCT03070119)。ASOs的问题是,由于分子在下游工作以停止蛋白质合成,如果找到成功的分子,可能需要重复剂量来抵消成人ALS患者活性基因新转录的mRNA。
2021年6月FDA批准Apic-Bio的靶向SOD1基因突变型ALS的AAV基因治疗APB-102的IND申请。APB-102是一款利用重组AAVrh10载体,可表达抗SOD1微小核糖核酸(miRNA),再通过miRNA抑制SOD1 mRNA合成,从而减少SOD1突变蛋白的产生,缓解患者症状。AveXis也采用了使用rAAV9载体表达抗SOD1的发夹RNA(shRNA)的方法。
2023年3月13日,Anew Medical公司宣布了其领先的分泌型Klotho(s-KL)候选基因疗法在ALS的严格动物模型中的临床前体内研究结果:通过s-KL基因、肌肉靶向AAV载体和肌肉特异性启动子,在肌肉和中枢神经系统中过量表达s-KL蛋白,可以增强运动功能,产生更多的神经肌肉接头,减少氧化应激和神经炎症。产生更高的主要肌肉群的动作电位,诱导改善肌肉力量和肌肉质量,减少谷氨酸诱导的脊髓兴奋性毒性,推迟疾病的发生,并与未治疗的对照组相比提高总生存率。
2023年4月25日,渤健/lonis的反义寡核苷酸疗法Qalsody(Tofersen)获FDA加速批准上市,用于治疗超氧化物歧化酶1 (SOD1)突变所致的肌萎缩侧索硬化(ALS)患者。Tofersen是首款针对ALS的基因靶向疗法,可与编码SOD1的mRNA结合,使其被核糖核酸酶降解,从而减少SOD1蛋白的产生。但根据临床试验研究结果显示,接受Tofersen治疗的患者人群在临床肌萎缩侧索硬化功能评定量表(ALSFRS-R)相较基线下降较少(p=0.97),该研究不具有统计学意义。
由于ALS只是多种基因相关神经退行性疾病中的一种,与其他类似疾病如脊髓性肌萎缩症的比较表明,ALS的基因治疗是可能的。在I型和II型SMA中,ASO、nusinersin (Spinraza)和通过AAV9载体的基因治疗AVXS-101 (zolgensma)现在是金标准疗法,zolgensma在单一静脉剂量下有效,如果在两岁前治疗,这些患者可以正常生活。然而,在ALS中,单剂量疗法不足以阻止疾病,因为ASOs不会影响RNA的新转录,并且由于已经受影响的神经元数量,通过基因疗法靶向患病神经元将是困难的。
此外,静脉内和鞘内给药将具有挑战性,因为对于目前需要静脉给药的ALS药物,如依达拉奉(Radicava),患者需要多次前往输液中心,或者如果他们的保险允许,通过port-a-cath进行家庭治疗。随着ALS疾病负担的加重,患者可能会面临交通困难和到达输注位置的时间困难,从而降低他们的整体生活质量。
目前ALS相关研究虽然已经步入临床治疗阶段为单基因突变患者提供个性化的ASOs治疗策略有可能成为ALS的常规治疗方案。但是由于ALS遗传学是高度动态的,表现为多种表型,在治疗ALS的过程中需要同时考虑功能丧失(LOF)和功能获得(GOF),因为一些LOF效应可以显着改变疾病的相关表型。虽然治愈ALS仍然任重道远,但随着学界不断合作与努力,一个又一个新的治疗会不断出现,期待在不久的将来能够有更多的基因治疗药物开展临床试验,帮助到更多的患者。
参考文献
-
Fang T, et al. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells. 2022 Jun 29;11(13):2066.
-
Shefner JM, et al. Amyotrophic Lateral Sclerosis Clinical Trials and Interpretation of Functional End Points and Fluid Biomarkers: A Review. JAMA Neurol. 2022 Dec 1;79(12):1312-1318.
-
Korobeynikov VA, et al. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat Med. 2022 Jan;28(1):104-116.
-
Brenner D, et al. Update on genetics of amyotrophic lateral sclerosis. Curr Opin Neurol. 2022 Oct 1;35(5):672-677.
-
Feldman EL, et al. Amyotrophic lateral sclerosis. Lancet. 2022 Oct 15;400(10360):1363-1380.
-
Mead RJ, et al. Amyotrophic lateral sclerosis: a neurodegenerative disorder poised for successful therapeutic translation. Nat Rev Drug Discov. 2023 Mar;22(3):185-212.

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





谢谢分享
37